






CLIP (UV-crosslinking and immunoprecipitation)是研究蛋白质和RNA 相互作用的重要技术,它利用了蛋白质和RNA 在256 nm 紫外光照射下会发生共价交联的特性。但是CLIP 是较难掌握的一种技术。由于紫外交联的效率很低(1%~5%),交联的RNA 含量很少,RNA容易降解,RNA 连接效率低,实验流程复杂(~100步) 等因素都增加了应用CLIP 的难度。
为了提高紫外交联效率,科学家开发了PAR-CLIP ( photoactivatable – ribonucleoside – enhanced CLIP) 技术,可以将交联效率提高100~1 000 倍。这个方法中最重要的一点就是依赖了具有光活性的核糖核苷类似物,例如4-硫尿核苷,在活体细胞中将其插入到新生RNA转录本中。在365nm紫外灯辐射的细胞中,光活性的核糖核标记的RNA被诱导与RBP相互作用。免疫共沉淀所要的RBP,然后分离被交联和共沉淀的RNA。RNA随即被转换成cDNA文库并且进行深入测序。当使用4-硫尿核苷时,交联的序列将会产生T-C的转变,因此通过PAR-CLIP所准备的cDNA文库能够准确的定位交联的位置。
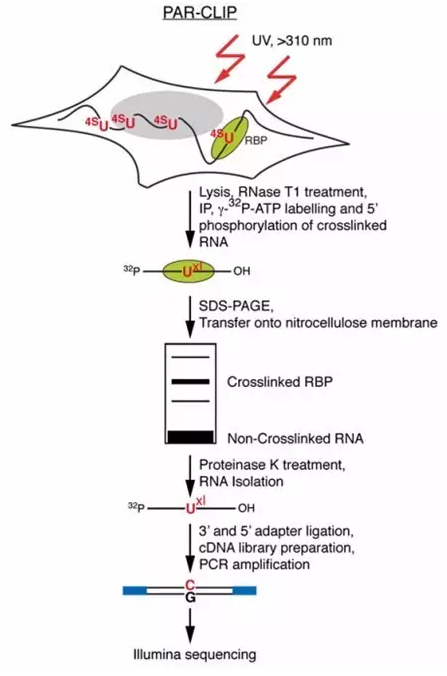 PAR-CLIP 流程图(Methods Mol Biol. 2016;1361:77-90)
PAR-CLIP 流程图(Methods Mol Biol. 2016;1361:77-90)
具体实验方法
★
试剂准备
1. 4-硫尿核苷储存液 (1 M): 260.27 mg 4-硫尿核苷, 1 ml DMSO
2. 多西环素储存液(10 mg/ml): 10 mg 多西环素, 1 ml DMSO
3. 5× NP40 裂解液: 制备无DTT和蛋白酶抑制剂的5×的储存液, 50 mM HEPES (pH 7.5), 150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% (v/v) NP40
4. 柠檬酸盐-磷酸盐缓冲液: 4.7 g/L 柠檬酸, 9.2 g/L Na2HPO4, pH 5.0
5. IP洗涤缓冲液: 50 mM HEPES-KOH (pH 7.5), 300 mM KCl, 0.05% (v/v) NP40
6. 高盐洗涤缓冲液: 50 mM HEPES-KOH (pH 7.5), 500 mM KCl, 0.05% (v/v) NP40
7. 脱磷酸缓冲液: 50 mM Tris-HCl (pH 7.9), 100 mM NaCl, 10 mM MgCl2 and 1 mM DTT
8. 磷酸酶洗涤缓冲液: 50 mM Tris-HCl (pH 7.5), 20 mM/review/reagent/EGTA.html EGTA, 0.5% (v/v) NP40
9. 多核苷酸激酶 (PNK)缓冲液无DTT: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl2
10. PNK 缓冲液: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl2, 5 mM DTT
11. SDS-PAGE loading buffer: 10% glycerol (v/v), 50 mM Tris-HCl (pH 6.8), 2 mM EDTA, 2% SDS (w/v), 100 mM DTT, 0.1% bromophenol blue
12. 2×蛋白酶K缓冲液: 100 mM Tris-HCl (pH 7.5), 150 mM NaCl, 12.5 mM EDTA, 2% (w/v) SDS
13. 5× NP40 裂解液、IP洗涤缓冲液、高盐洗涤缓冲液等在实验前直接加入0.5 mM DTT和无EDTA的蛋白酶抑制剂cocktail (Roche) 后使用。
★
实验步骤
01
细胞培养和体外交联
1) 在培养皿中培养增殖细胞(15cm 培养板,需20-50块板),培养至饱和度为80%。
2) 直接加入4-硫尿核苷至终浓度100μM至细胞培养液中。
3) 在交联后14小时 ,每块板中用10ml预冷的1×PBS洗一次细胞,然后完全移除PBS。
4) 将培养板放在有冰的托盘上,并且在0.15 J/cm2、365nm的紫外灯下进行照射。
5) 在培养板中加1ml1×PBS,然后用橡胶棒将细胞刮下,将细胞转移到50ml离心管中,4℃下500g离心5min,去除上清液。
02
RNA酶T1消化
1) 在沉淀的细胞中加入3倍体积的1×NP-40裂解缓冲液,并且在冰上静置10min。
2) 将细胞裂解液在13,000g、4℃下离心15min,弃沉淀。
3) 用0.2um膜注射器式滤器进一步清洗细胞裂解液。
4) 加RNA酶T1\至终浓度1U/μl并且在22℃水浴锅中孵15min。然后在冰上使其反应5min。
03
预备磁珠
1) 每毫升细胞裂解液加入10μ蛋白 G 磁珠颗粒到1.5ml小试管中。用1ml柠檬酸盐-磷酸盐缓冲液清洗珠子两遍。
2) 按照原来珠子混悬液的体积再用两次同体积的柠檬酸盐-磷酸盐缓冲液重悬。
3) 每毫升悬液加0.25 μg特异性抗体,然后在室温进行40min旋转孵育。
4) 用1ml柠檬酸盐-磷酸盐缓冲液洗珠子两次。
04
免疫共沉淀
1) 加入20μl新配置的抗体-磁珠处理细胞裂解液(用RNA酶T1处理),然后在4°C下15ml离心管中旋转混匀1h。
2) 用磁力分选器收集磁珠,并转入到1.5ml小离心管中。
3) 用IP缓冲液清洗珠子3次
05
第二次RNA酶T1消化以及去磷酸化
1) 加入RNA酶T1至终浓度100 U/μl,然后在22°C水浴锅中孵育珠子混悬液15min,接着在冰上静置5min。
2) 用高盐缓冲溶液洗珠子三次。
3) 用等体积的去磷酸缓冲液重悬珠子。
4) 加入小牛小肠碱性磷酸酯酶至终浓度0.5 U/μl,然后在37°C下静置混悬液10min。
5) 用1ml磷酸盐缓冲液洗珠子两次
6) 在不含有DTT的多核苷酸激酶中洗珠子两次。
7) 用原来体积的珠子PNK缓冲液重悬珠子。
06
标记RNA片段并交联
1) 加γ-32P-ATP至终浓度0.1 μCi/μl和T4多核苷酸激酶至终浓度1 U/μl到上面的珠子混悬液。37°C下静置混悬液30min。
2) 加入无放射性标记的ATP到终浓度100μM,然后再在37°C下静置混悬液5min。
3) 用800μl不含有DTT的多核苷酸激酶的缓冲液洗珠子5次。
4) 在65μl十二烷基磺酸钠-聚丙烯酰胺(SDS-PAGE)加样缓冲液中重悬珠子。
07
电泳、洗脱和消化
1) 将放射线标记的混悬液在95℃加热器上加热5min(使其变性并释放同交联的RNA的免疫共沉淀的RBP),然后涡旋。
2) 在磁力分选器上将磁珠移除,并将上清液转移到新的1.5ml的小试管中。
3) 每个孔道中加40μl Novex Bis-Tris 4-12%预处理过的上清液,然后跑聚丙烯酰胺凝胶(200V、45-60min)。
4) 将胶从胶槽中拿出,将胶放在一个平板上。为了使胶上的片段在磷光指示带上被标出,我们植入三个小的放射性的胶段到胶三个角中。放射性胶段能从胶中收集,被最先用来纯化放射性标记的合成寡聚核苷酸。
5) 将胶包起来放置在空白的磷光板上扫描1h使其完全显现。
6) 将胶摆在磷光板上面,使用植入的胶段来用作定位。将预计的RBP大小片段的胶带切下,然后转移到透析柱中,加入800μl 1×SDS电泳缓冲液。
7) 在1×SDS电泳缓冲液中、100V下电洗脱交联的RNA-RBP混合物2h使其从胶中分离出来,切断胶条然后在D型管中电洗脱。
8) 加等体积的2×的蛋白酶K缓冲液到电洗脱物中,然后在加蛋白酶K至终浓度1.2mg/ml。55℃静置30min。
9) 用酸性的苯酚/氯仿/碘乙酰胺(25:24:1,pH=4.0)来还原RNA,紧接着用氯仿萃取。加1μl糖原(10mg/ml)和3倍体积的乙醇来沉淀RNA。用10μl水来溶解沉淀。
08
cDNA库和高通量测序
富集下来的RNA加3’接头,5’接头,然后逆转录后,PCR扩增获得从DNA文库,使用高通量测序技术进行测序并进行生物信息学分析。
参考文献:
1. PAR-CLIP (Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation): a step-by-step protocol to the transcriptome – wide identification of binding sites of RNA-binding proteins.Methods Enzymol. 2014;539:113-61 .
2. Mapping the Transcriptome-Wide Landscape of RBP Binding Sites Using gPAR-CLIP-seq: Experimental Procedures.Methods Mol Biol. 2016;1361:77-90.
来第一个抢占沙发评论吧!