







- 背景
RNA在生命进程中发挥着非常重要的作用,我们对RNA最初的认识是中心法则中的mRNA,它负责将DNA包含的信息传递给直接参与调控生命活动的主要承担者—蛋白质。然而在RNA世界中,大部分是非编码RNA,包括LncRNA,rRNA, tRNA, snRNA和microRNA,它们在调控生命进程中也发挥着重要作用。
值得一提的是:这些非编码RNA都是线性分子从5’开始到3’结束,在众多的非编码RNA中,存在一种特殊的共价闭合环状RNA分子—circRNA。从1976年首次在植物病毒中发现至今,CircRNA让我们对于RNA的功能又有了新的认识,目前对于CircRNA的认识也只是冰山一角,但是从最近的研究来看,CircRNA已经掀起一股新的研究热潮。
对于研究CircRNA来说,要解决的第一个问题就是先找到CircRNA。已经有相应的软件可以对CircRNA进行预测,比如find_circ、CIRCexplorer、CIRI,但是不同方法的差异非常大。这些CircRNA检测工具主要用于检测不含polyA的RNA测序数据或者用RNaseR处理过后的RNA测序数据。
因为CircRNA在经过富集之后可以很容易的被检测出来。然而,在做科学研究的时候,基于节约经费考虑,一般都是做total RNA测序,这些数据运用更多,信息量更大。因此,急需一个基于Total RNA测序结果能够准确预测CircRNA的工具。在这种情况下,UROBORUS应运而生。
- UROBORUS预测CircRNA的主要步骤
准备工作:在运行UROBORUS.pl之前,电脑需要先装好下面三个软件
1、TopHat
2、Samtools
3、Bowtie
用TopHat工具将RNA reads mapping到基因组上,得到unmapped.sam文件。
UROBORUS以unmapped.sam作为输入数据。从unmapped.sam文件的reads两端提取长度为20bp的片段,形成一个末端配对的fastq文件(R20 1.fastq andR20 2.fastq)然后再用TopHat与人类参考基因组比对(默认允许两个碱基的错配)。
上面的比对结果可以得到两类连接的reads,Balanced Mapped Junction(BMJ) reads和Unbalanced Mapped Junction(UMJ) reads。BMJ reads指的是两端有至少20bp的碱基可以和两个back-spilced外显子的连接区配对,UMJ reads指的是一端有少于20bp的碱基可以和两个back-spilced外显子的连接区配对。
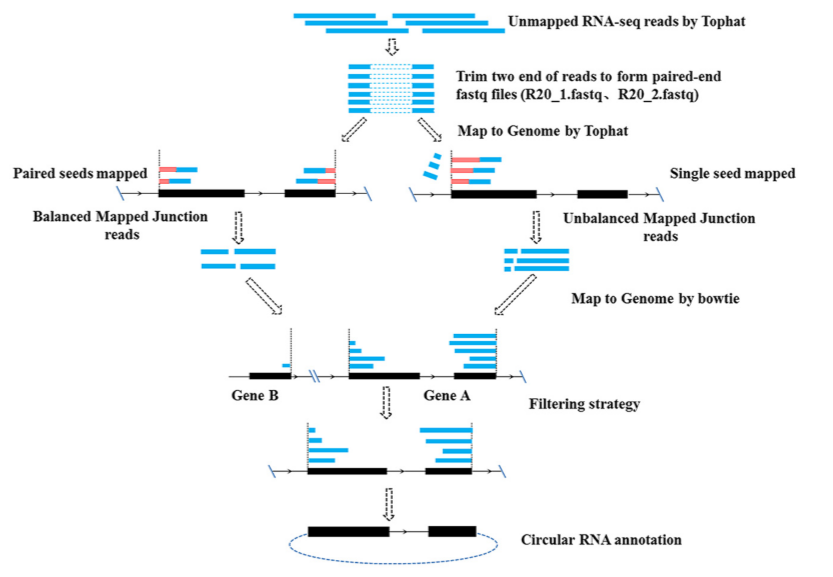
图1:UROBORUS预测CircRNA主要步骤简介
对UROBORUS软件进行综合测评
UROBORUS运用实例
选取了7个少突胶质细胞瘤,20个恶性胶质瘤和19个正常人的脑组织的Total RNA数据(去除rRNA)按照上面的操作步骤,分别预测了其中的CircRNA,结果发现:预测的结果中有已经被证实的CircRNA(circularHIPK3, CAMSAP1, MAN1A2, FBXW4, REXO4, ZKSCAN1, ZBTB44,FAM120A, MAP3K1, ZBTB46,NUP54, RARS, CRKL and XPO1)。
选取两个CircRNA,它们被UROBORUS鉴定出来的可能性。图2表示可能找到的跨越 Circular ERC1的14和15个外显子之间连接点的BMJ 和 UMJ reads,有30条明显跨越连接点的BMJ reads,但是却有43条只比对到Circular ERC1边界的UMJ reads。
另外,Circular CORO1C连接点的只有7条UMJ reads而不含BMJ reads,这表明只用BMJ reads的软件鉴定不到circular CORO1C,而 UROBORUS选取了BMJ和UMJ reads会使得它在检测CircRNA是更灵敏。
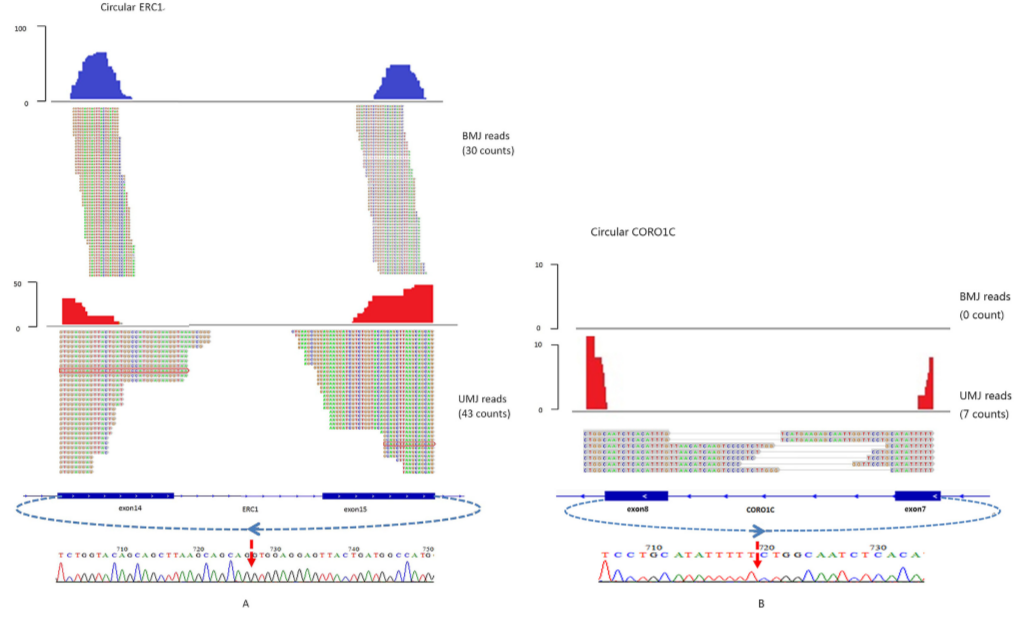
图2:Circular ERC1能被鉴定到的BMJ和UMJ reads,Circular CORO1C能能被鉴定到的UMJ reads
UROBORUS假阳性的概率
为了检测软件的假阳性概率,一般选择运行含有PolyA的RNA数据(不含CircRNA),理论上是不会有发现CircRNA的,如果有那就是假阳性。
用同样的方法,我们检查了UROBORUS假阳性的概率,在输入样本大小为127万条reads时,检测出2条reads 对应的一个CircRNA,假设所有已报道的CircRNA是假阳性的,UROBORUS的假阳性概率为每一百万reads约0.79。因此,预测UROBORUS的FDR < 0.013。
UROBORUS与find_circ和CIRCexplorer的比较
用这三个软件分别预测了前面的样本,根据表1的数据可知UROBORUS能预测到的CircRNA比find_circ多,但是少于CIRCexplorer,由于预测CircRNA的策略不同,CIRCexplorer在第一步的时候忽略了UMJ reads对应的CircRNA,并且过高的估计了能检测到的CircRNA的表达水平,它之所以能预测到更多的CircRNA是由于检测偏差造成的。
find circ会漏掉很多spanning reads很短的CircRNA和低估了它们的表达水平,这样就造成检测的到的CircRNA更少了。
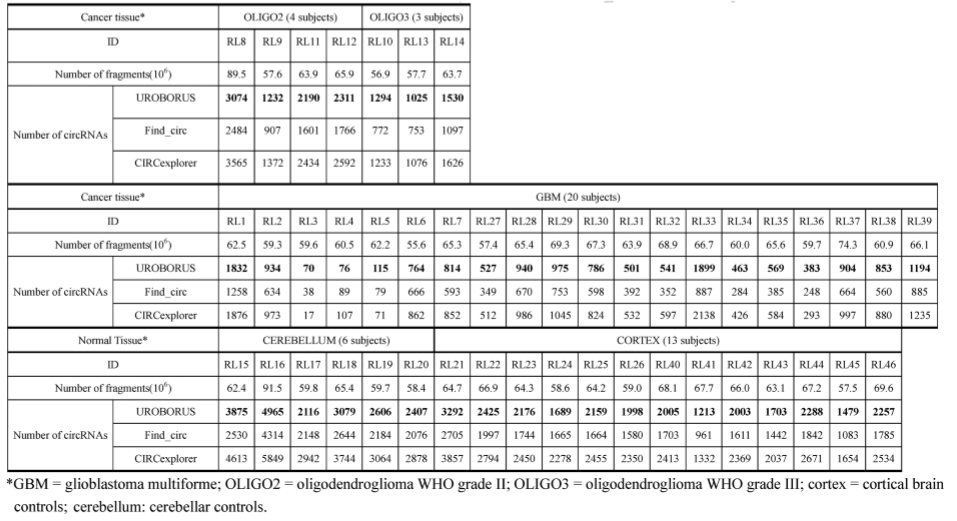
表1:由UROBORUS、find_circ和CIRCexplorer鉴定到的CircRNA的数目
用Venn图表比较三个软件鉴定到的CircRNA,其中307个CircRNA是共有的,find circ能够鉴定出196与CIRCexplorer和 UROBORU不同的circRNAs;CIRCexplorer能够鉴定出 524与find circ和UROBORUS不同的circRNAs,UROBORUS 能够鉴定出 726 与find circ和CIRCexplorer不同的circRNAs。
这表明 UROBORUS 能够检测到更多新颖的circRNAs,基于它们不同的mapping和筛选策略,这三个软件可以当做互补的工具。并且在检测高表达的circRNA时,这3款软件效果类似。在运行上,UROBORUS占用的最大内存为6.3G,比另外两个大一点,同时运行10G的RNA测序数据时,UROBORUS要消耗4.8 h。
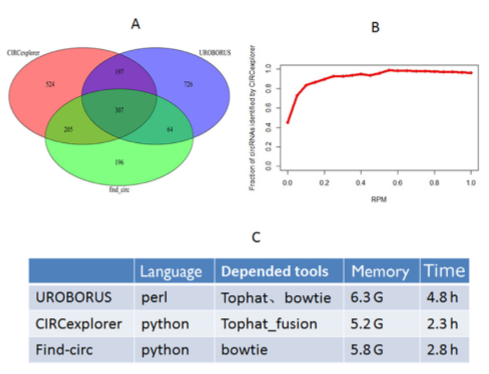
图3:A 用Venn图比较了三种软件鉴定到的CircRNA。
B当CircRNA检测高表达水平提高时,UROBORU和CIRCexplorer鉴定到了更多相似的CircRNA。
C 比较三种方法运用的语言、依赖的工具、内存和运行时间。
总结
UROBORUS是一款基于Total RNA测序数据预测全转录组CircRNA的软件。
与CIRCexplorer和 find circ相比,UROBORUS在预测Total RNA中表达较低的CircRNA时准确率更高,使用起来也更加方便。
find circ所使用的是linux操作系统,对于生物学家来说要掌握这个并不容易,并且用find circ预测得到的CircRNA的数量明显少于CIRCexplorer和UROBORUS。CIRCexplorer是TopHat融合的修正,它分两步的mapping策略,第一步mapping会得到一个重叠区的索引,然后把所有没有与基因组mapping上的序列与第一步叠区的索引比对。
这样就必然造成忽略了UMJ reads对应的CircRNA,并且过高的估计了能检测到的CircRNA的表达水平。UROBORUS可以避免掉这些误差。
尽管,三个软件通过不同的策略寻找CircRNA,然而,当CircRNA表达水平提高时, UROBORUS能找到更多的普通的和高表达的CircRNA,更重要的是,当CircRNA表达水平很低时,UROBORUS也能精确的预测到,这点是findcirc 和CIRCexplorer所无法达到的。
本软件不足之处在于,只能检测外显子之间连接产生的CircRNA,不能预测出内含子之间连接和基因间隔区连接形成的CircRNA。
尽管内含子之间连接和基因间隔区连接形成的CircRNA只占总CircRNA的一小部分,然而我们在为下一代的UROBORUS研发而努力,希望可以检测到更多内含子之间连接形成的CircRNA。
下载文献:circular-rna-profile-in-gliomas-revealed-by-identification-tooluroborus
来第一个抢占沙发评论吧!