







作为我们星球上最古老的生物大分子,早在DNA产生之初,RNA可能就扮演现在储存遗传信息的功能,RNA自身还能折叠成复杂的高级结构,并能与蛋白结合参与精确而复杂的生理调节。
RNA在生物信息传递过程中发挥至关重要的作用,在中心法则中RNA是联系物种遗传信息与蛋白的纽带。与此同时在RNA生物体内的种类也纷繁复杂,在人类基因组中序列除5-10%的mRNA以外,绝大多数的RNA是无法编码蛋白的ncRNA,它们包括snRNA,siRNA,piRNA,miRNA,rRNA,tRNA,circRNA,lncRNA, snoRNA,antiRNA,telomerase RNA等。其中部分siRNA,miRNA, piRNA,lncRNA, circRNA在调控基因表达及染色质结构中发挥重要作用。
siRNA通常是一段长21nt的双链RNA,在植物中通过与靶标的RNA特异性结合降解RNA导致基因沉默;miRNA则是一段长约20-24nt的单链RNA,它在进化上高度保守,通过翻译机制抑制基因的表达。piRNA(piwi-interacting RNA)是一类长约24-32nt的ncRNA,主要存在于生殖系统,初步推测其可能在通过结合Piwi蛋白在沉默基因转录过程,维持生殖细胞和干细胞功能,调节翻译和mRNA稳定性中发挥功能。lncRNA是一类长度超过200nt的ncRNA,在染色质重塑,转录调控,转录后调控及蛋白代谢水平调节细胞的基因表达,从而调控生物个体的发育过程,疾病发生和环境应答等生命活动;circRNA是一类不具有5`cap和3`ployA,以共价键形成环形结构的ncRNA分子,部分circRNA含有miRNA应答元件,可充当竞争性内源RNA(competing endogenousRNA,ceRNA),在胞浆中以类似海绵一样吸附miRNA,进而解除miRNA对其靶基因的抑制作用,从而调节基因的表达。一部分含内含子的环状RNA(EIcirRNA)一般无法出核,存在细胞核内通过顺式作用调控基因的转录。
RNA通过自身的序列结构或结合到特定的蛋白中参与靶标DNA或RNA的调节或改变染色质结构,从而实现其生物学功能,那对识别特定RNA的所结合的蛋白、RNA、DNA就成为了一个重要的科学问题。
随着二代测序的发展,对全基因组RNA不同转录本的识别已经变得越来越容易,根据不同的实验目的结合二代测序技术来研究RNA结合事件的技术被不断开发出来。在下文我们将分成RNA-Protein\RNA-RNA\RNA-DNA三部分来介绍该领域的最新进展。
一、RNA-Protein研究
1. RNA Pull Down Assay(1)
为了鉴定特定的RNA分子是否与某一种特定的蛋白结合,研究人员根据DNA-Protein实验中DNA Pull down的实验方法进行改进,使其在RNA上实现。
技术步骤如下:
(1)设计末端携带T7 RNA Promoter的引物扩增DNA片段;
(2)利用T7 RNA polymerase 进行转录,转录过程添加携带生物素的dNTP,使转录本携带生物素;
(3)裂解细胞核,回收核或胞浆内容物;
(4)将转录获得的RNA与内容物进行孵育;
(5)用磁珠捕获RNA-蛋白复合物;
(6)用western blot或蛋白质谱鉴定特定的蛋白。
该方法主要用于鉴定某一种RNA结合的特定的RNA binding蛋白是否具有结合或在不同分化周期及细胞周期蛋白的结合情况。
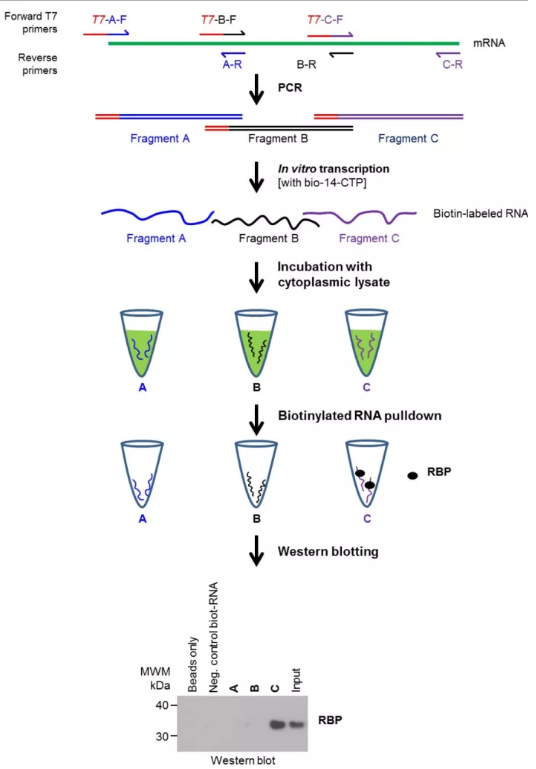
图1.RNA Pull Down 示意图
2. REMSA(RNA Electrophoretic Mobility Shift Assay, REMSA)
电泳迁移率检测是一种用来研究蛋白与其结合的DNA或RNA互作的技术,其原理是蛋白结合了核酸后其分子质量变大,相对于free probe而言,核酸-蛋白复合物在聚丙烯酰胺凝胶上迁移变慢,通过检测迁移率可以鉴定核酸是否与蛋白结合。
实验步骤如下:
(1)利用T4 PNK 和32P标记DNA或RNA,并纯化;
(2)EMSA凝胶配制;
(3)抽提核裂解液或特定蛋白;
(4)核裂解液与核酸进行孵育,同时设置对照实验(探针冷竞争反应,突变探针的冷竞争反应,Super-shift反应);
(5)电泳检测。
3. RIP(RNA Binding Protein Immunoprecipitation,RNA结合蛋白免疫沉淀)
该技术用来研究特定蛋白与全基因组RNA结合情况的技术。该技术主要借鉴了ChIP-seq的蛋白抗原抗体特异性结合的原理,利用特定的抗体将RNA-蛋白复合物进行富集,并用磁珠进行捕获,回收特定的RNA进行逆转录并测序。
其技术步骤如下:
(1)细胞核裂解;
(2)抗体孵育,免疫沉淀;
(3)回收RNA;
(4)cDNA合成,添加测序接头,纯化;
(5)二代测序;
(6)生物信息学数据分析;
(7)功能验证。
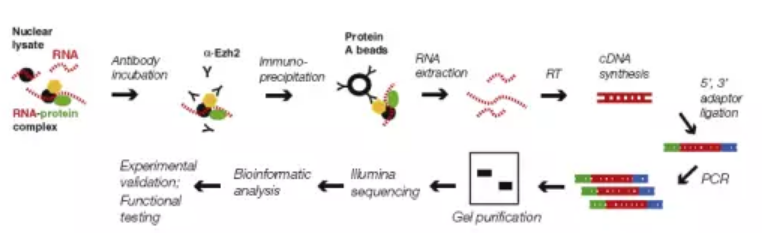
图2.RIP流程图
4. CLIP(ultraviolet cross-linking immunprecipitation,紫外交联免疫沉淀)
为了识别蛋白与RNA分子结合特定的位点,Jernej Ule发明CLIP技术。该技术主要技术原理是利用紫外交联的方法将RNA于蛋白特定在结合位点交联在一起,利用抗体富集蛋白结合的RNA,并用RNase消化掉未被蛋白包裹的区域,保留RNA-蛋白结合的核心区域。最后回收RNA进行建库测序。
其主要步骤有:
(1)用紫外照射细胞,使蛋白和RNA分子进行交联反应;
(2)裂解细胞,用目的蛋白抗体进行免疫共沉淀;
(3)利用核酸酶RNase对RNA进行切割,保留核心区域;
(4)alkaline 磷酸酶去除RNA末端的磷酸基团,防止下一步自连;
(5)用RNA连接酶在3`端连接上RNA接头;
(6)用T4PNK 磷酸激酶对RNA 5`端进行磷酸化,添加32p;
(7)SDS-PAGE分选蛋白-RNA复合物;
(8)解交联并添加接头,进行cDNA合成;
(9)PCR扩增,并测序;
(10)生物信息学分析。
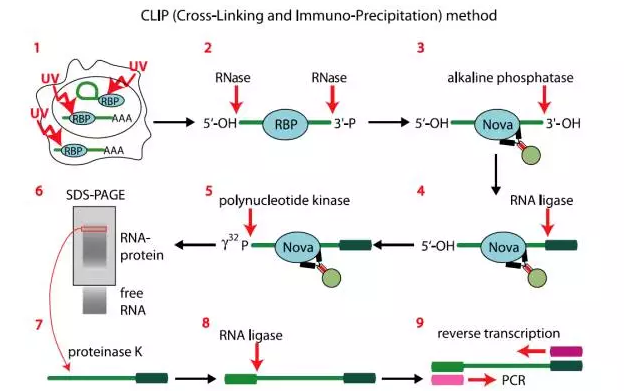
图3.CLIP流程图
5. Ago HITS-CLIP (high-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation)
前面已经提到miRNA可以与mRNA结合调控基因的表达,有研究指出miRNA仅需要6-8nt与mRNA精确配对即可发挥作用。但这对利用碱基组成重头预测靶标 mRNA是一个巨大的挑战。miRNA与Argonaute蛋白复合物构成RISC核心元件,并引导Argonaute锚定到特定的mRNA上,Argonaute蛋白具有内切核酸酶活性,在二价金属离子的辅助下可以切割mRNA分子,在动物细胞的miRNA沉默途径中,Argonaute蛋白也可以不依赖内切酶活性来达到基因沉默的作用,它通过抑制靶标mRNA翻译的方法,以及诱导mRNA发生去腺苷酸化后降解的方式来抑制基因表达。洛克斐勒大学霍华德休斯研究所的Robert B. Darnell研究组在高通量CLIP共价交联Argonaute-RNA复合物,通过分析RNA的组成,来确定miRNA-RNA全局互作图谱,作者在这篇文章中重点分析小鼠神经细胞中miR-124全局的互作图谱,并得到20中最常见的miRNAs的互作图谱。该方法可以用于鉴定组织特异性的miRNA-mRNA调控网络。
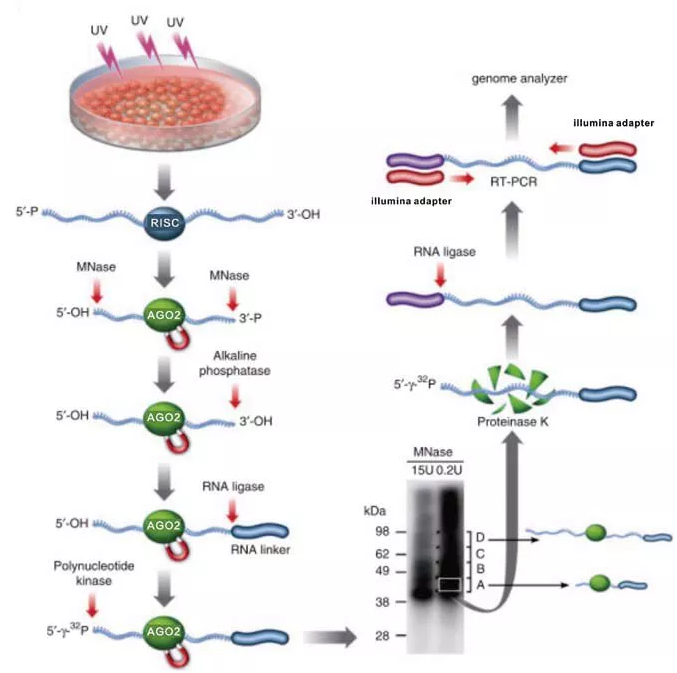
图4.Ago HITS-CLIP示意图
其步骤如下:
(1)紫外交联细胞;
(2)裂解细胞,提取核裂解物;
(3)用Ago抗体进行孵育;
(4)用MNase消化RNA;
(5)用Alkaline phosphalase磷酸酶去除RNA3`末端的磷酸基团;
(6)用T4 PNK磷酸激酶将5`OH转换成磷酸基团;
(7)用SDS-聚丙烯酰胺凝胶切胶;
(8)回收RNA,并在5`端添加RNA接头;
(9)RT-PCR生成cDNA分子;
(10)测序及生物信息学分析;
(11)功能分析及鉴定。
6.PAR CLIP (Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation)
RNA翻译是非常重要的科学问题,RNA结合蛋白(RBPs)和小RNA-核糖核蛋白复合物(miRNPs对真核生物基因表达起到了很重要的作用,这些蛋白跟DNA结合蛋白一样,可以与相关的序列结合来指导它们的表达。这些RBPs和miRNPs共同来调控细胞内许多过程,包括mRNA的成熟、运输、修饰和翻译。CLIP虽然可以用来鉴mRNA与核糖蛋白的结合,但是UV虽然可以使得RNA与蛋白直接交联,但交联效率较低,且未被交联的分子更容易被逆转录,导致了最终的结果又很强的噪声,很难分辨出那些是直接交联的RNA。
洛克菲勒大学的Thomas Tuschl课题组在CLIP的基础进行了改进,早期的CLIP选用254nm的紫外线照射,他们用更低能量的356nm紫外来照射细胞。而取而代之的是在细胞培养体系中添加了具有光活性的核糖核苷类似物,例如4-硫尿核苷,在活体细胞中将其插入到新生RNA转录本中。在低能量的紫外照射过程中,光活性的核糖核标记的RNA被诱导与RBP相互作用。并在RT过程中U会被转换成C,使得在最终的文库中可以特定地识别到高精度单碱基水平的蛋白和RNA结合。
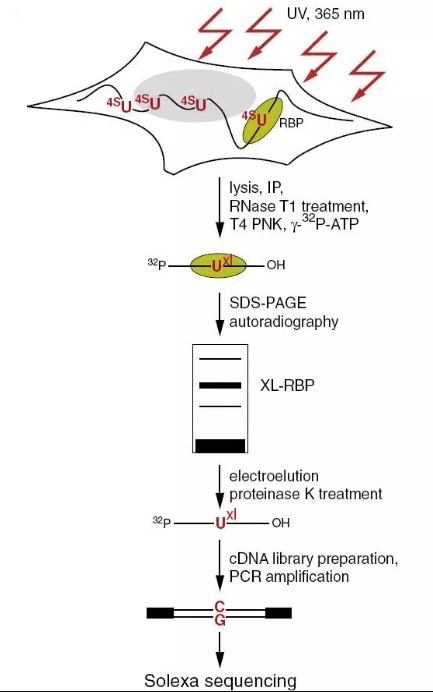
图5.PAR-CLIP流程图
7.eCLIP(enhanced CrossLinking and ImmunoPrecipitation followed by high-throughput sequencing)
在对CLIP-seq文库进行分析发现,其PCR冗余较高,一个文库需要测序上百million的数据才能获取到足够的有效数据,加州大学Gene W. Yeo对原有的方法进行了改进,他们在添加3`端的接头上加了可以用来识别不同拷贝的index,同时改进RT的步骤顺序,在蛋白解交联之前进行RT实验,使得逆转录在蛋白结合位点终止,从而可以精确识别到蛋白的结合位点,获取了单链DNA后,它在DNA3`端再添加DNA的接头,从而可以进行PCR扩增,用于二代高通量测序。该技术使得循环数大大减低,有效数据量显著增高。
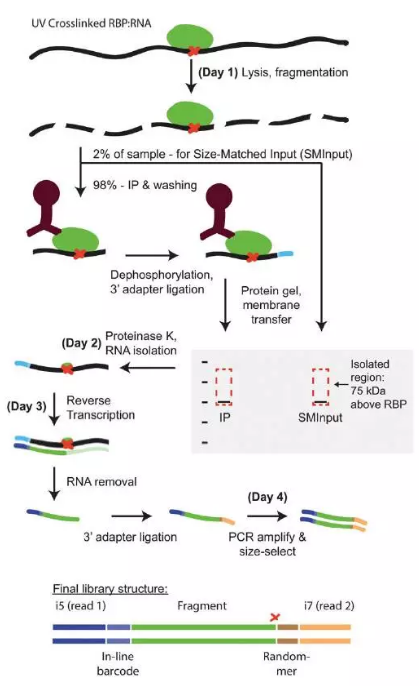
图6.eCLIP流程图
二、RNA-RNA互作研究
1. CLASH(crosslinking,ligation, and sequencing of hybrids)
RNA-RNA互作在众多的生物学进程中被证实,几乎所有的RNA都必须有正确的折叠才能发挥功能,而不同的RNA分子之间的结合,构成了RNA代谢的诸多途径,包括 mRNA前体的剪切,核糖体合成,以及通过miRNA调控mRNA的稳定性。
如核仁小分子RNA(snoRNA)是一类广泛分布于真核生物细胞核仁的ncRNA,具有特别保守的结构元件,并以此划分为3大类:box C/D snoRNA、box H/ACA snoRNA和MRP RNA。其中boxC/D和boxH/ACA是已知snoRNA的主要类型,以碱基配对的方式分别指导着核糖体RNA的甲基化和假尿嘧啶化修饰。研究表明,snoRNA在rRNA的生物合成中发挥作用之外,还能够指导snRNA、tRNA和mRNA的转录后修饰。此外,还有相当数量的snoRNA功能不明,被称为孤儿snoRNA(orphan snoRNA)。snoRNA 与rRNA的结合被普遍报道,大多数的box C/D snoRNA基因通过甲基化转移酶Nop1对rRNA进行甲基化,但是与之相反,U3 snoRNA可以配对到rRNA前体的多个位点,这些互作被认为可以协助rRNA前体的正确折叠,且对rRNA前体的加工是必须的。但是这些互作的证据都是间接的,为了获得更直接的证据,David Tollervey课题组发明了一种新的方法CLASH。
他们在CLIP的基础上直接将两段不同的RNA连接成Chimera,从而在文库中被识别出来。但是在他们的文章中也报道连接成嵌合片段的reads仅占整个文库的不到1%。同时rRNA占整体的RNA占据了80%以上,因此很难获得lncRNA-mRNA的互作情况。
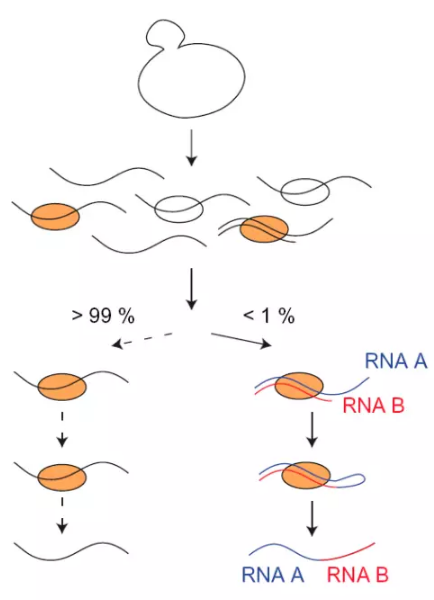
图7.CLASH示意图
2. Mapping RNA interactome in vivo (MARIO)
鉴于CLASH有效数据少,迫切需要有新的技术来实现嵌合片段的富集,加州大学的Sheng Zhong研究组,借鉴TCC的方法,在蛋白上进行生物素化,使整个反应在磁珠上进行,在RNA于RNA进行连接步骤中他们用添加了一带有生物素的link,使得最终的嵌合片段可以被富集,从而大大提高了数据的利用率。
主要实验步骤分为:
(1)紫外线共价交联RNA-蛋白;
(2)蛋白的生物素化;
(3)与链亲和素结合,将RNA-Protein固定到磁珠上;
(4)连接带有生物素的link;
(5)在dilute环境下进行RNA连接;
(6)消化蛋白,用RNase酶部分消化RNA;
(7)用链亲和素富集带有生物素的RNA;
(8)生成cDNA;
(9)二代测序及生物学分析。
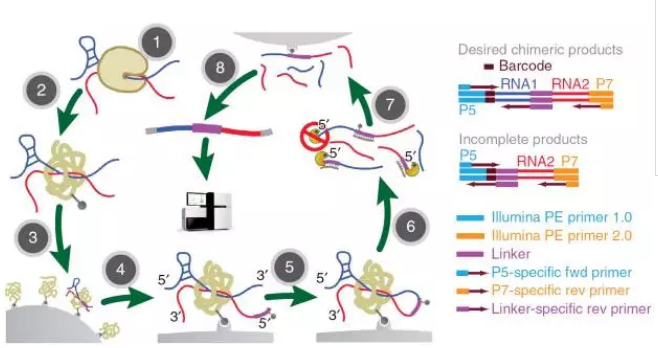
图8.MARIO流程图
三、RNA-DNA互作研究
在X染色体失活过程中,长非编码RNA XIST会随机地结合到一条X染色体中,有研究证实Xist的结合与PRC2和H3K27me3成线性相关,暗示其与PRC2共同弥漫到基因组中,使其结构变得异常紧密(10),有研究证实CTCF能结合到XIST的启动子上抑制基因的表达,而另一个非编码RNA JPX是剂量敏感的激活基因,它能特异性地结合CTCF,能类似于海绵一样从XIST的启动子上吸附CTCF,从而激活Xist表达(11)。有越来越多的证据表明lncRNA对染色质的构象的形成发挥关键的作用。因此如何鉴定RNA与DNA的直接互作已经成为了迫切的需求。
1.R3C(12)
kcnq1pt1,是一个长达91kb的非编码RNA,它被认为是一个能在父本的染色体影响其反义基因kcnq1的印迹表达。Kcnq1ot1 RNA是一个双向抑制子,能顺式调控超过1Mb的Kcnq1 印迹位点的基因。但对于它的扮演的角色并不是很清楚。吉林大学的He Zhang设计了一个全新的实验R3C, 他们先将RNA 反转录成DNA,然后在cDNA第二链的生成中,掺入生物素,使其可以被链亲和素进行富集。
实验步骤如下:
(1)甲醛交联DNA-RNA-Protein复合物;
(2)cDNA生成第一链;
(3)第二链生成中,掺入生物素;
(4)限制性内切酶进行酶切;
(5)DNA连接酶连接反应;
(6)蛋白解交联;
(7)设计特定的引物进行扩增,测序。
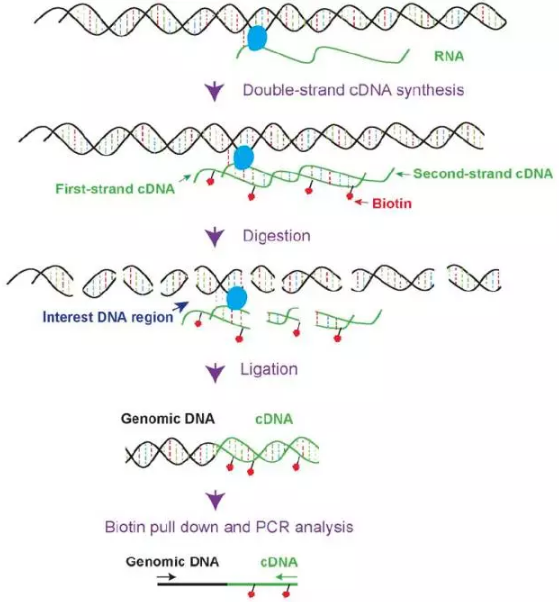
图9.R3C示意图
3. GRID-seq(in situ global RNA interactions with DNA by deep sequencing)
R3C只能用来研究特定RNA与DNA互作情况,并不能获得全局的RNA-DNA的互作情况,付向东实验室在今年发表的一项新技术(GRID-seq)解决了这一问题
其实验步骤主要分为两部分:
第一部分在体内反应:
(1)用甲醛、DSG双重交联细胞核;
(2)用Alu限制性内切酶进行酶切;
(3)在RNA原位连接一段双链的DNA-RNA link,link中携带生物素;
(4)RNA反转录;
(5)去除游离的linker;
第二部分在体外反应:
(6)cDNA与DNA进行连接;
(7)解交联,用链亲和素磁珠捕获携带生物素的DNA;
(8)从磁珠中洗脱DNA,合成cDNA另一条链;
(9)因linker中携带Mme1酶切位点,因此可以剪切DNA片段,保留近65碱基的DNA片段;
(10)切胶回收DNA,添加测序接头PCR扩增用于二代测序。
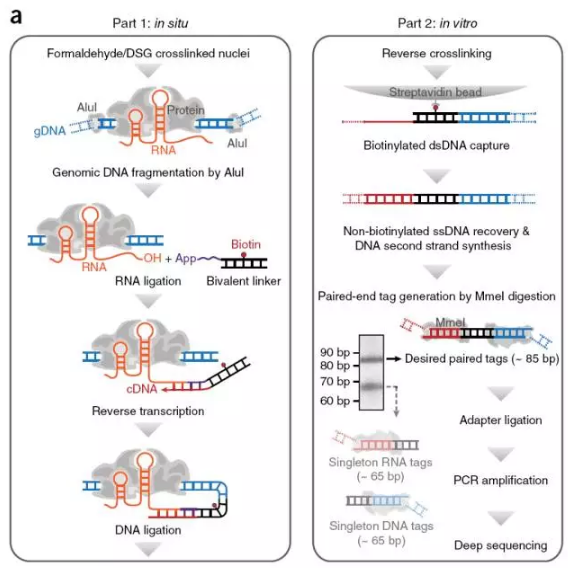
图10.GRID-seq流程图
四、DNA-RNA-Protein互作研究
1.ChIRP(Chromatin Isolation by RNA Purification)
RNA调控染色质的机制可能不仅仅是通过裸露的RNA分子,可能由各种转录因子及RNA结合蛋白的参与,在一次实验中同时鉴定出RNA-DNA-蛋白复合体的组成成分,显然能极大地帮助我们理解其调控的机制。Howard Y. Chang 实验室开发的一门技术ChIRP,很好地解决了这一难题。
实验步骤如下:
(1)对RNA-DNA-Protein复合物进行交联;
(2)超声打断DNA分子;
(3)设计特定lncRNA的检测探针,探针中携带便于捕获的生物素;
(4)探针与复合物进行孵育;
(5)用磁珠捕获复合物;
(6)用RNaseH和RNaseA对RNA进行消化,分别回收基因组DNA和RNA结合蛋白;
(7)测序及质谱鉴定DNA和蛋白的组成。
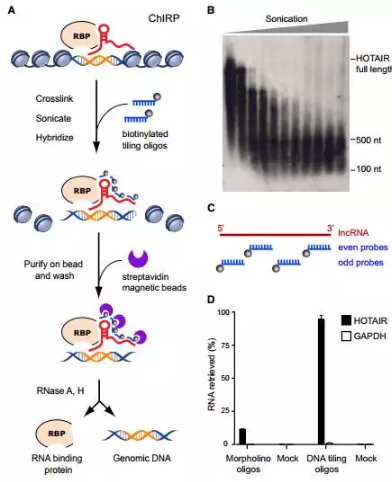
图11.ChIRP流程图
因缘际会,整理RNA互作研究的知识脉络,对该领域的研究进展有了一次比较全面的认识和介绍,该领域是一个非常前沿的领域,由于技术的难度,很多工作可谓是十年磨一剑,如阮一骏老师的RiCH-PET技术,很多年前就得知已经开发了,囿于个人认识和能力所限,谬误在所难免,读者权当是科普便好。如果有兴趣深入研究,小编也在文章后列了一系列参考文献,可参照阅读,谬误之处,敬请斧正,在此致谢。
(1)de Silanes I L, Zhan M, Lal A, et al. Identification of a target RNA motif for RNA-binding protein HuR[J]. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101(9): 2987-2992.
(2)Parsley T B, Towner J S, Blyn L B, et al. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase[J]. Rna, 1997, 3(10): 1124-1134.
(3)Zhao J, Ohsumi T K, Kung J T, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq[J]. Molecular cell, 2010, 40(6): 939-953.
(4)Ule J, Jensen K B, Ruggiu M, et al. CLIP identifies Nova-regulated RNA networks in the brain[J]. Science, 2003, 302(5648): 1212-1215.
(5)Chi S W, Zang J B, Mele A, et al. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps[J]. Nature, 2009, 460(7254): 479-486.
(6)Hafner M, Landthaler M, Burger L, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP[J]. Cell, 2010, 141(1): 129-141.
(7)Van Nostrand E L, Pratt G A, Shishkin A A, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP)[J]. Nature methods, 2016, 13(6): 508-514.
(8)Kudla G, Granneman S, Hahn D, et al. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast[J]. Proceedings of the National Academy of Sciences, 2011, 108(24): 10010-10015.
(9)Nguyen T C, Cao X, Yu P, et al. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO[J]. Nature communications, 2016, 7.
(10)Simon M D, Pinter S F, Fang R, et al. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation[J]. Nature, 2013, 504(7480): 465-469.
(11)Sun S, Del Rosario B C, Szanto A, et al. Jpx RNA activates Xist by evicting CTCF[J]. Cell, 2013, 153(7): 1537-1551.
(12)Zhang H, Zeitz M J, Wang H, et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus[J]. J Cell Biol, 2014, 204(1): 61-75.
(13)Li X, Zhou B, Chen L, et al. GRID-seq reveals the global RNA-chromatin interactome[J]. Nature biotechnology, 2017, 35: 940.
(14)Chu C, Qu K, Zhong F L, et al. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions[J]. Molecular cell, 2011, 44(4): 667-678.
文章来源: 三维基因组Magic(微信公众号)

来第一个抢占沙发评论吧!