







ABSTRACT
Circular RNAs (circRNAs) represent a new type of regulatory noncoding RNA that only recently has been identified and cataloged. Emerging evidence indicates that circRNAs exert a new layer of post-transcriptional regulation of gene expression. In this study, we utilized transcriptome sequencing datasets to systematically identify the expression of circRNAs (including known and newly identified ones by our pipeline) in 464 RNA-seq samples, and then constructed the CircNet database (http://circnet.mbc.nctu.edu.tw/) that provides the following resources: (i) novel circRNAs, (ii) integrated miRNA-target networks, (iii) expression profiles of circRNA isoforms, (iv) genomic annotations of circRNA isoforms (e.g. 282 948 exon positions), and (v) sequences of circRNA isoforms. The CircNet database is to our knowledge the first public database that provides tissue-specific circRNA expression profiles and circRNA–miRNA-gene regulatory networks. It not only extends the most up to date catalog of circRNAs but also provides a thorough expression analysis of both previously reported and novel circRNAs. Furthermore, it generates an integrated regulatory network that illustrates the regulation between circRNAs, miRNAs and genes.
INTRODUCTION
Circular RNAs (circRNAs) are a family of RNAs whose head 3′ and tail 5′ ends covalently bond together to result in a circular form. Identified more than thirty years ago, circRNAs were first hypothesized to be relics of pre-cellular evolution (1) with the phenomenon of the 3′ and 5′ ends covalently joining called ‘exon scramble’ (2) or ‘back-spliced junctions’. In plant cells, circRNAs were known to be pathogenic and deemed viroid (3,4). In contrast, circRNAs found expressed in mammal cells (5–8) were generally considered to be rare products of splicing error.
In recent years, high throughput sequencing technology has dramatically expanded the scope of transcriptomics research (9) and made it possible to more accurately investigate the expression of non-coding RNA genes. In 2012, Salzman et al. (10,11) developed an algorithm to detect scrambled exons in RNA-seq datasets, and reported that circular RNA isoforms are actually predominant in many human gene isoforms. By 2013, Memczak et al. (12) developed an improved version of the algorithm and found that some circRNAs serve as natural microRNA ‘sponges’. The reliability of the back-splicing junction detection algorithm can be validated through a protocol developed by Jeck et al. (13), which includes examining RNA-seq samples treated with RNase R (14). For instance, a circRNA named CDR1as (15) expressed in human and mouse brain was shown to negatively regulate miR-7 in a post-transcriptional manner (12,15,16); this mechanism appears to be evolutionarily conserved (12). Extended identification of circRNAs in mouse (17), fly (18) and other animals (19) suggests that circRNA ubiquity is evolutionally-conserved. Further evidence indicates that human circRNA expression exhibits tissue specificity, and now tens of thousands of circRNAs have been found and reported across human tissues (17,20–25). CircRNAs functional correlation with brain function, synaptic plasticity (26,27) and fetal development (28) has recently been reported. The fact that cell free circRNAs were found stable in saliva (29) as well as exosomes (30) makes circRNA a promising diagnosis biomarker.
Given the emerging understanding of the biological importance of circRNAs and research efforts to understand them, we constructed a database called CircNet to extend the catalog of reported circRNAs and provide resources to aid in their study. Previously reported and newly identified human circRNAs are cataloged, with CircRNA expression metadata in the form of a heatmap illustrating circRNA expression profiles across 464 human transcriptome samples additionally provided. Embracing the idea that circRNAs are enriched with conserved miRNA binding sites (31) and function as natural miRNA sponges (12), CircNet maps circRNA–miRNA–mRNA interactions into regulatory networks. An integrated genome browser illustrates the position of circRNAs in the human genome, and relative locations of repeat sequences are presented to address the issue of circRNA biogenesis as the biogenesis of circRNAs correlates to repeats in the flanking area (13,32). A provided search function enables the exploration of CircNet. Users can choose an interested gene or miRNA to access its associated network, expression profile, and genome position simultaneously in CircNet. Overall, CircNet provides interactive tools for users to easily access comprehensive information regarding expression profiles across many conditions, genome loci, close repeat sequences, post-transcription regulation networks, and references to previous studies for circRNAs.
DATA COLLECTION AND DATABASE CONTENT
CircNet's data collection, cataloguing and analysis are summarized in Figure Figure1.1. The process started from identifying back-spliced junction sites in the collected datasets to find a total of 212 950 circRNAs, including 53 687 novel RNAs. From them, we defined 34 000 high-confidence circRNAs by selecting back-spliced junction sites with supporting experiments ≥3. To associate circRNA isoforms with miRNAs, mature human miRNA sequences and experimentally verified miRNA target genes were acquired from miRBase (33) and miRTarBase (34). Perfect alignments of 6mer, 7mer-A1, 7mer-m8 and 8mer lengths between miRNA target seeds and circRNA isoforms were collected in CircNet, where 6mer represents the match of position 2 to 7; 7mer-A1 represents a 6mer alignment plus one A in position 1; 7mer-m8 represents a matched from position 2 to 8; and 8mer represents match of all 1 to 8 position (35). Appearance frequency of these miRNA target sites were normalized by isoform length. From the distribution of this frequency across all transcripts, including linear isoforms of the genes, only those isoforms with a P value less than 0.001 are reported to sponge miRNA in CircNet.
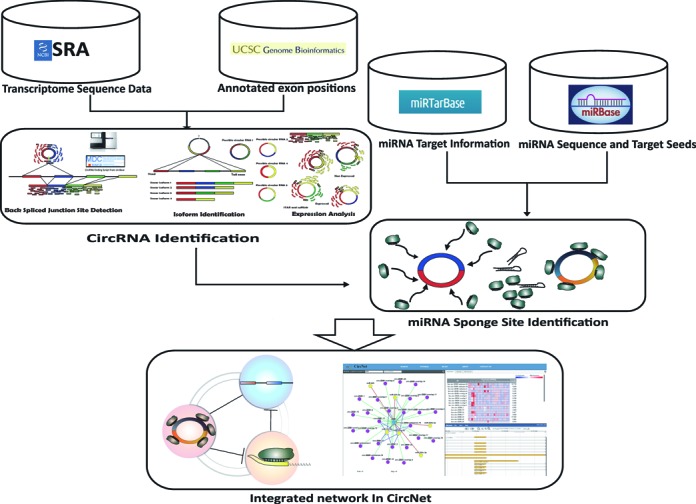
Figure 1.
Framework of the database construction in CircNet. The graph illustrates how the network in CircNet was constructed. For circRNA identification, transcriptome sequencing data sets were obtained from the NCBI Sequence Read Archive (SRA). The back-spliced ...
Data collection
Reported human back-spliced junction sites were also collected from the supplemental information provided in other work (10–13,20–25,27). In addition, 464 RNA-seq samples were collected from a wide range of independent experiments across 26 human tissues and 104 disease conditions from the NCBI Sequence Read Archive (36). These samples cover research topics ranging from detecting gene variation (37–41) to searching for long non-coding RNAs (42). Datasets used in recent publications (17,20–25) regarding circRNA identification were also included. CircRNA back-spliced junction sites were identified within these samples. To acquire the expression patterns of circRNAs within these samples, we designed a pipeline to perform expression profiling.
CircRNA identification
CircRNA back-spliced junction sites in the collected datasets were identified using the algorithm proposed by Memczak et al. (16), which has also been used by the circBase database to detect back-spliced junction sites (43). As shown in Figure Figure1,1, reads spanning junction sites that cannot be fully aligned onto the reference genome were analyzed to identify back-spliced junction sites. Junction sites meeting the same threshold criteria in Memczak et al. (16) were cataloged in CircNet.
To acquire the full length nucleotide sequence from RNA-seq reads, back-spliced junction sites were compared with the hg19 human genome annotation as obtained from UCSC genome browser (44). Given recent research, we recognized that multiple circRNA isoforms might originate from the same back-spliced junction site (11,26). Based on the method proposed by Salzman et al. (11), we identified multiple circRNA isoforms with annotated transcripts if the ‘head’ and ‘tail’ positions of a detected back-spliced junction were located exactly at the position of the exon junction sites belonging to the same annotated transcripts. Since back-spliced junction site flanking exons could potentially be composed of multiple isoforms, we included all the possible isoforms in the annotation for the expression analysis and miRNA target search. In addition, we found many reported and novel back-spliced junction sites that did not align exactly to well-annotated exon locations. For those circRNAs associated with these back-spliced junction sites having small misalignments to exon locations, flanked exons and a small portion of intron sequence were considered as parts of the isoforms. Still other junction sites were found to be located in intergenic positions while others, despite overlapping with certain genes, localized to their antisense strands. For these circRNAs with no apparent flanked exons, we took the entire flanked sequence between the ‘head’ and ‘tail’ position of the back-spliced junction sites for expression analysis and miRNA target search. Given the possibility of flanking intron areas as part of the circRNAs as described in Gao et al. (20), we took the full flanked sequences into consideration for expression and miRNA target search. The process is illustrated in Supplementary Figure S1. Expression profiling was performed through integrating the tool Cufflinks (45,46) with STAR (47). Expression level of the isoforms can also be estimated through FPKM (Fragments Per Kilobase of exon per Million fragments mapped) values during the process.
The newly identified circRNAs were named by following the nomenclature suggested by Jeck et al. (48). The ID of a circRNA specifies the gene symbol of the source gene. If the back-spliced junction site is not exactly aligned with annotated exons, an ‘overlap’ tag is included in the ID. Similar tags suggesting that the circRNA is antisense to the origin gene are included in the ID for the applicable circRNAs.
MicroRNA sponge detection
Potential miRNA binding sites on circRNAs were identified through iteratively searching the circRNA isoform sequences for miRNA target sequences deemed typical: 6mer, 7mer-A1, 7mer-m8 and 8mer sequences (35). Perfect complementarity was required of these sequences for a circRNA–miRNA target relationship to be identified. To normalize the number of occurrences of these sites, the below formula was used:
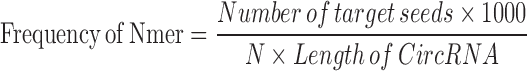
equation M1
With this formula, four frequency numbers can be acquired from each pair of circRNA and miRNA. To distinguish circRNA from linear isoforms, frequency values were also calculated for linear mRNA and miRNA pairs. P-values for each circRNA and miRNA relationship were acquired by one minus the calculated the culminated distribution function of the frequency Z score. The circRNA–miRNA pair with P-value < 0.001 represents high regulatory potential between the circRNA and miRNA. Information of these regulatory relationships was combined with target genes of miRNA from miRTarBase (34) to form the networks demonstrated on CircNet.
WEB INTERFACE
The web interface of CircNet is summarized in Figure Figure2.2. The main page of CircNet is composed of three panels and a search box (top left). With the search box, users identify a gene or miRNA of interest to first find its associated gene–miRNA-circRNA regulatory network (left panel). If a gene is queried, this identifies the post-transcriptional regulatory relationships of (i) miRNAs targeting the given gene and (ii) circRNAs originating from within the given gene. If instead a miRNA is searched for, circRNAs predicted to serve as sponges of the given miRNA are summarized in the network. Clicking on a circRNA within the network view re-centers the network on the selection (Figure (Figure2C).2C). To change the degree of stringency of predicted relationships, the ‘Support’ drop down menu in the top left corner allows users to restrict the view to only high-confidence circRNAs, with confidence based on having back-spliced junction sites found in multiple studies or been found to pass the thresholds defined Memczak et al. (16) in multiple RNA-seq experiments. In the default setting, only those circRNAs whose back-spliced junction sites found in three or more sources are shown.
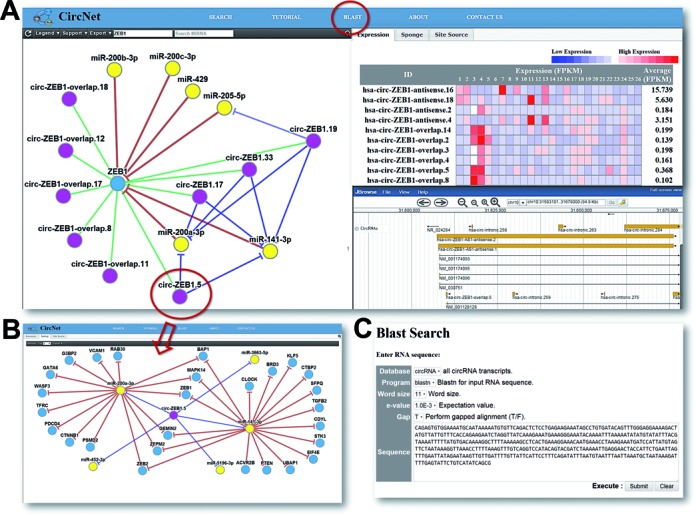 Figure 2.
Figure 2.
Overview of CircNet web interface.
(A) Users can search a gene or miRNA of interest, and then CircNet generates three panels as follows:
(1) the left panel: a gene–circRNA–miRNA regulatory network,
(2) the top-right panel: circRNA expression ...
The top-right panel consists of three sub-categories: ‘Expression’, ‘Sponge’ and ‘Site Source’. These categories change simultaneously when the user clicks on one of the circRNA nodes on the network in the left panel. In the ‘Expression’ tab, a heat map of the expression profile for all the circRNAs as well as overlapped linear transcripts illustrated on the network across all 464 RNA-seq samples is demonstrated. In the ‘Sponge’ tab, an extended network focused on the selected circRNA, all miRNAs sponged by the selected circRNA, and their associated target genes is shown. An example of this kind of extended network is illustrated in Figure Figure2B.2B. In the ‘Site Source’ tab, a table shows experiments in which multiple back-spliced junction site reads were found. Links to the peer-reviewed source reports of the junction sites can also be found in this table; the longer the list, the stronger the evidence supporting the existence of this back-spliced junction site.
The bottom-right panel synchronizes with the user search query to generate a genome browser view localizing to the search target. The browser currently contains three tracks: (i) Reference sequence: the Hg19 human genome sequence, (ii) HG19_repeats: any repeat sequences on Hg19 human genome sequence for the searched subject; and (iii) CircRNAs: all circRNAs as well as overlapped linear transcripts around the selected area.
Sequence search is also available on CircNet (Figure (Figure2C).2C). Through integrated BLAST, users can search for all circRNAs that align with input nucleotide sequences. Back-spliced junction sites overlapped with only intergenic regions can also be accessed through this function.
EXAMPLE APPLICATIONS
We use the ZEB1-derived network as an example to illustrate the information provided by CircNet (Supplementary Figure S2). When ZEB1 is inputted as the keyword, CircNet collects available information about miRNAs targeting ZEB1, circRNAs originating from the ZEB1 sequence, and the regulatory relationship between the circRNAs and miRNAs to create in an integrated mRNA-miRNA-circRNA regulatory network. The network suggests that the circRNAs circ-ZEB1.5, circ-ZEB1.19, circ-ZEB1.17 and circ-ZEB1.33 have the potential to sponge the miRNA miR-200a-3p. Since miR200a has been reported to target ZEB1 (34), this suggests the existence of a negative feedback control loop. The integrated genome browser provides the visualization of these circRNAs: all these circRNAs contain the exon in position chr10:31791276–31791437, which is enriched with miR-200a targeting seeds. From the flanking area of this specific exon, we found the flanking MIR repeat sequences MIR_dup1543 and MIR3_dup768 and MIR3_dup769, providing a lead to the biogenesis of the circRNA. As illustrated in Supplementary Figure S3, expression patterns provided in CircNet indicate that all four identified circRNAs — circ-ZEB1.5, circ-ZEB1.19, circ-ZEB1.17 and circ-ZEB1.33 — were up-regulated in available normal lung tissue samples compared to the lung cancer samples.
DISCUSSION AND FUTURE DEVELOPMENT
Regarding other circRNA databases, Glazar et al. constructed circBase (http://circbase.org/) (43), and Li et al. constructed starBase (http://starbase.sysu.edu.cn/) (49). Supplementary Table S1 lists the comparison between CircNet, starBase and circBase and it suggests some strong advantages of CircNet in comparison to these other works. One of the advanced features that makes CircNet distinctive is the novel design of circRNA naming in the catalog. Emerging evidence suggests that multiple circular isoforms originate from the same back splice junction site (11,26). Given this, we catalog circRNAs based on distinctive isoforms instead of regarding one observed back-spliced junction position as a single circRNA. Following the Jeck et al. suggestions in 2014 Nature Reviews (48), we designed a circRNA naming system which provides information regarding source genes, whether the circRNA is antisense or intronic, and whether the back splice junction site is exactly located on well annotated exons. This annotation and incorporation into an integrated genome browser should provide a clear method for future curation Expression profile and accessibility to the examined sample condition provided in CircNet would be a useful tool to study circRNA tissue specific function as well as correlation to disease. One observation worth mention is the potential wide existence of double negative feedback control loops in the network. As seen in the ZEB1-derived network example, the circRNA originating from within ZEB1 was putatively found to sponge a miRNA targeting ZEB1. The double negative control relationship on ZEB1 gene and mir-200 family was previously reported by Bracken et al. in 2008 (50). The correlation between mir-200 ZEB1 regulation and lung adenocarcinoma was recently reported by Yang et al. (51). Data provided in CircNet indicates that circRNAs spanning the chr10:31791276–31791437 exon of ZEB1 play significant roles in this regulation. With the newly discovered ZEB1 circRNAs enriched with mir-200 targeting seeds, a hypothetical double negative feedback control loop of circRNA is demonstrated on the default network on homepage of CircNet. Further inquiries using the regulatory networks identified using CircNet may discover additional novel feedback loops with applicability to human disease.
AVAILABILITY
The CircNet database will be continuously maintained and updated. The database is now publicly accessible at http://circnet.mbc.nctu.edu.tw/.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENTS
The authors would like to thank the Ministry of Science and Technology of the Republic of China for financially supporting this research. We also thank the UST-UCSD International Center of Excellence in Advanced Bio-engineering sponsored by the Taiwan Ministry of Science and Technology I-RiCE Program, Veterans General Hospitals and University System of Taiwan (VGHUST) Joint Research Program, and MOE ATU. We would like to thank The Cancer Genome Atlas for use of data.
FUNDING
Ministry of Science and Technology of the Republic of China [MOST 101-2311-B-009-005-MY3, MOST 103-2628-B-009-001-MY3, MOST 103-2320-B-005-004, MOST 104-2621-M-005-005-MY3 and MOST 104-2320-B-005-005]; UST-UCSD International Center of Excellence in Advanced Bioengineering sponsored by the Ministry of Science and Technology I-RiCE Program [MOST 103-2911-I-009-101]; Veterans General Hospitals and University System of Taiwan (VGHUST) Joint Research Program [VGHUST103-G5-11-2]. MOE ATU. Funding for open access charge: Ministry of Science and Technology of the Republic of China, Taiwan [103-2628-B-009-001-MY3]. This work was supported by a research grant from Health and welfare surcharge of tobacco products, Ministry of Health and Welfare (MOHW103-TD-B-111-08; MOHW104-TDU-B-212-124-005)
貌似永久不能用了
同意