








Profiling expression of coding genes, long noncoding RNA, and circular RNA in lung adenocarcinoma by ribosomal RNA-depleted RNA sequencing
用去除核糖体RNA(rRNA-depleted)的RNAseq技术研究肺腺癌mRNA, lncRNA, circRNA表达谱
期刊:FEBS Open Bio 影响因子:2.143
发表单位:江苏省肿瘤医院放射科
文献解读:李纪伟,上海生命基因
1 摘要
非编码RNA在多种生物学过程、疾病包括肿瘤中发挥着重要作用。肺腺癌lung adenocarcinoma(简称LUAD),还没有进行过circRNA的系统研究。本研究,我们使用核糖体RNA去除的RNAseq技术对配对肺腺癌样品做了转录组表达谱研究包括编码基因、lncRNA, circRNA。测序得到的reads首先比对到人基因组分析mRNA和lncRNA。未能比对的reads使用circRNA预测算法鉴定候选circRNA。一共鉴定到1282个差异表达的编码基因。检测到19023个lncRNA,其中244个差异表达。AFAP1-AS1, BLACAT1, LOC101928245, and FENDRR 差异变化最高。大于2个backspliced reads的circRNA共鉴定到9340个,包括3590个新的circRNA,circRNA长度中值是530nt。circRNA丰度较低,超过一半的circRNA的backspliced reads <10。使用琼脂糖电泳和sanger法测序验证4个候选的circRNA,发现确实是环状的。本研究概述了肺腺癌mRNA,lncRNA,circRNA的表达谱。鉴定到9340个circRNA,提示circRNA在肺腺癌中广泛表达。
注:原始测序数据存储在GEO数据库,编号为GSE104854 。
2 材料方法
1 病人及组织样品
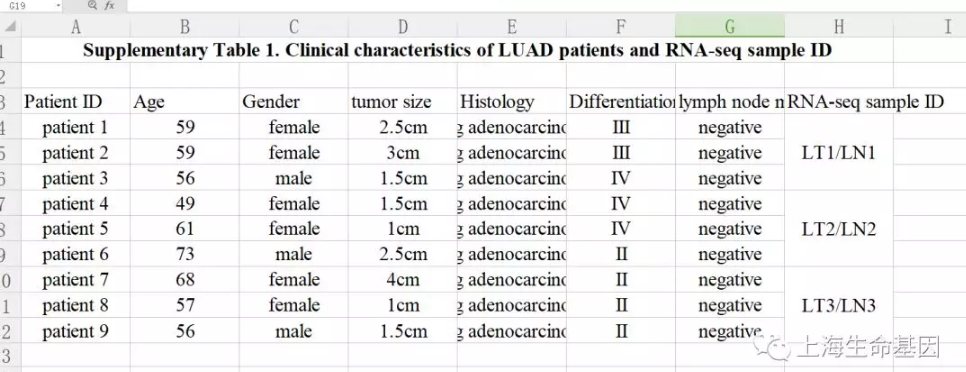
本研究获得江苏省肿瘤研究所伦理会的支持。配对的腺癌样品及癌旁样品来自于非小细胞肺癌(NSCLC)手术样品,时间是2012-2014,来源于江苏省肿瘤医院胸外科。由病理诊断丰富的病理医生判读配对的癌与癌旁样品。收集病人的临床和病理资料,同时获得病人的知情同意书。共收集9对肺腺癌样品,包括癌与癌旁组织。RNA样品混合测序共测得6个样品(3个癌组织,3个癌旁组织)。临床资料及RNA样品信息如Table S1。RNAseq的数据也已上传至GEO数据库。
2 细胞培养及PCR
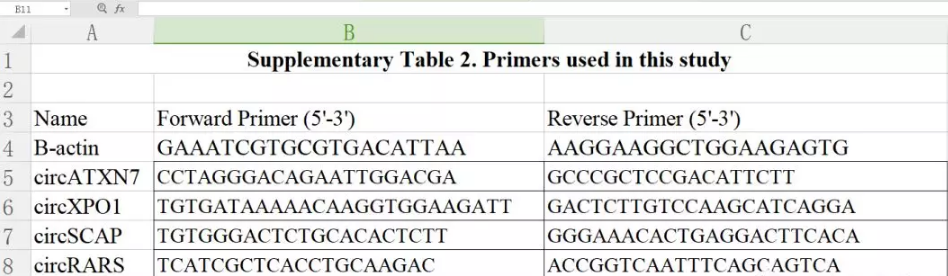
用RPMI 1640 (GIBCO, Invitrogen, Shanghai, China) 培养基培养A549细胞,培养基加入10% FBS (GIBCO-BRL, Invitrogen, Carlsbad, CA, USA), 100U.mL-1 penicillin, and 100mg.mL-1 streptomycin (Key-GEN),在空气潮湿,温度37 °C,5% CO2环境中培养。使用TRIzol (Invitrogen, Grand Island, NY, USA) 提取A549的细胞的totalRNA。使用PrimerScript RT Master Mix (Takara, Dalian, China)将1ug totalRNA在20ul体系中反转录。引物如Table S2。产物使用琼脂糖凝胶电泳分离并进行Sanger测序。
3 样品准备
使用TRIzol (Invitrogen, Shanghai,China)提取肺腺癌样品及癌旁样品total RNA。Bioanalyzer 2200分析RNA的完整性,并保存在-80℃。使用GeneRead rRNA Depletion Kit (QIAGEN, Venlo, Germany)去除rRNA。
4 文库构建及RNA测序
使用Ion Total RNA-Seq Kit v2.0 (Thermo Fisher Scientific, Inc., Waltham, MA, USA)准备单端cDNA文库。cDNA文库构建根据试剂盒说明书进行。测序仪器使用 proton sequencers(此仪器建库上机过程可查相关资料,并且此仪器目前很少使用,此处不多介绍)。
5 RNA测序及reads比对
对原始下机数据reads去除接头序列,去除“N”超过5%的序列,去除碱基质量值<13含量大于20%的reads。MAPSPLICE 软件 (v2.1.8) 将clean reads比对到人基因组(version: GRCh37),为了获得更多的可变剪切转录本,设置参数为(–s 22 –p 12 –ins 6 –del 6 –noncanonical)。
6 circRNA鉴定及定量
使用acfs流程https://code.google.com/p/acfs/鉴定circRNA, 主要包括以下几步:
1)获得未能比对到基因组的reads。使用BOWTIE2 version 2.2.5将reads比对到参考基因组[GRCH37.p13 NCBI]。
2)circRNA鉴定。未能比对的reads,使用BWA mem (bwa mem –t 1 –k 16 –T 20) 再次比对。部分比对的reads(a)两部分比对到同一个染色体上,距离不超过1MB;(b)同一条链;(c)比对方向相反,支持head-to-tail连接。将这类reads的连接点进一步用MaxEntScan33评估。根据受体与供体点的最高得分来确定准确连接点。支持head-to-tail连接点的reads数量≥2,连接点的得分值≥10。
3)表达量分析:为了得到circRNA的表达量。我们将unmapped reads使用 BWA mem 参数 (bwa mem –t 1 –k 16 –T 20) 重新比对到候选circRNAs。由于没有直接的证据能够确定circRNA的序列,我们通过中间存在的外显子注释获得。序列从5’端延伸到3’端以形成环状。能够比配到连接点的reads(至少重叠6nt)进行计数。
7 差异表达分析
使用DESeq算法过滤差异表达的基因,FC和FDR作为筛选标准,标准如下(a) FC > 2 or < 0.5; (b) FDR< 0.05。
8 GO富集分析
对差异表达基因(mRNA)进行GO富集分析,以了解基因参与生物学过程。首先下载GO注释信息,NCBI (http://www.ncbi.nlm.nih.gov/)、UniProt (http://www.uniprot.org/)、GO (http://www.geneontology.org/),将这些基因的注释信息进行整合,并作为背景信息。GO分析使用Fisher’s exact test计算p-value,使用FDR值对p-value进行校对。
9 通路分析
使用KEGG数据库进行通路分析以鉴定有意义的通路,同样使用Fisher’s exact test计算p-value,使用FDR值对p-value进行校对。FDR<0.05的认为有意义。
10 GO tree
GO tree是一种有向无环图,条目之间彼此关联,可以提供友好的数据整理和展示。我们挑选了有意义的GO条目(P-value<0.01)构建GO tree来了解实验影响的功能。
11 通路网络
KEGG数据库包含代谢、膜转运、信号转导、细胞周期通路。我们选择富集到的生物学过程的基因,使用CYTOSCAPE(version 3.2.1)进行通路网络化展示。
12 共表达网络
我们使用基因共表达网络,寻找基因之间的关系,基因共表达网络构建基于标准化的基因表达量。对于每两个基因,我们计算Pearson相关性系数,选择相关性高的成对基因构建网络。
在网络分析中,degree cetrality(度中心性,一个节点连接其他节点的数量为度)是最简单也是最重要的一种分析方法。度中心性是某个点与相连接点的数量。为了研究网络的权重,使用W-core理论来进行图形拓扑分析,W-core网络是一个子网络,在这个网络中节点至少与W个其他节点相连接。W-core网络是一个protein-protein网络,通常是紧密相关的蛋白。
网络分析的目的是为了确定核心调控因子,在一个网络中,核心调控因子与许多其他基因相连接,并有最高的度。考虑到不同的网络,核心调控因子要考虑在多个网络中存在,并具有较高的差异。
13 LncRNA-protein关系
使用NPInter(http://www.bioinfo.org/NPInter)建立lncRNA与蛋白质之间的网络,这个数据库包含了经过验证的noncoding RNA(except tRNA and rRNA) 与生物分子(proteins, RNA and DNA)的关系。这些关系是根据同行评审的论文人工收集的,许多来源于NONCODE (http://www.noncode.org/),
miRBase (http://www.mirbase.org/), and UniProt (http://www.uniprot.org/)。NPInter 根据ncRNA参与功能的过程对功能关系进行了分类。同时也提供高效的搜索功能,包括关系的搜索,相关论文及其他的信息。
3 结果
1 RNAseq概述
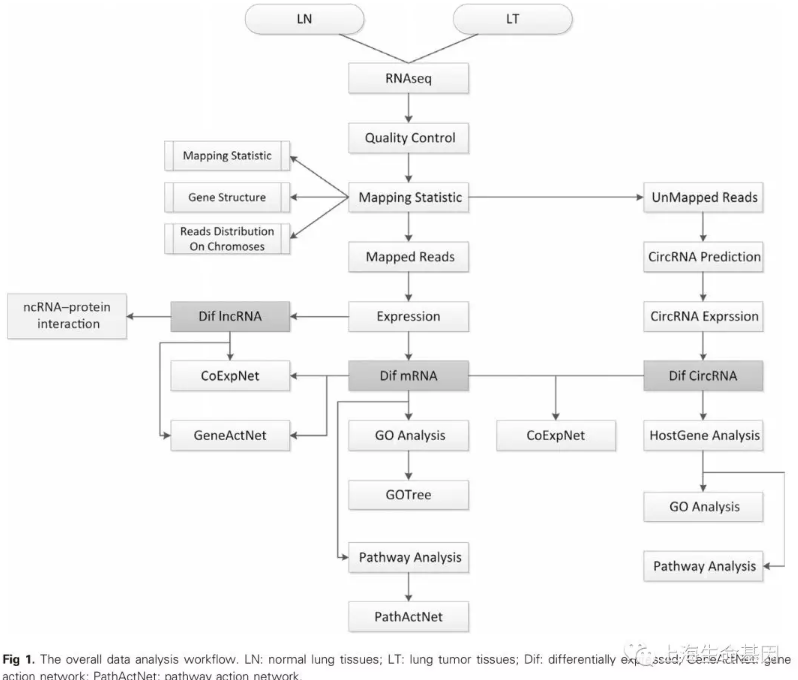
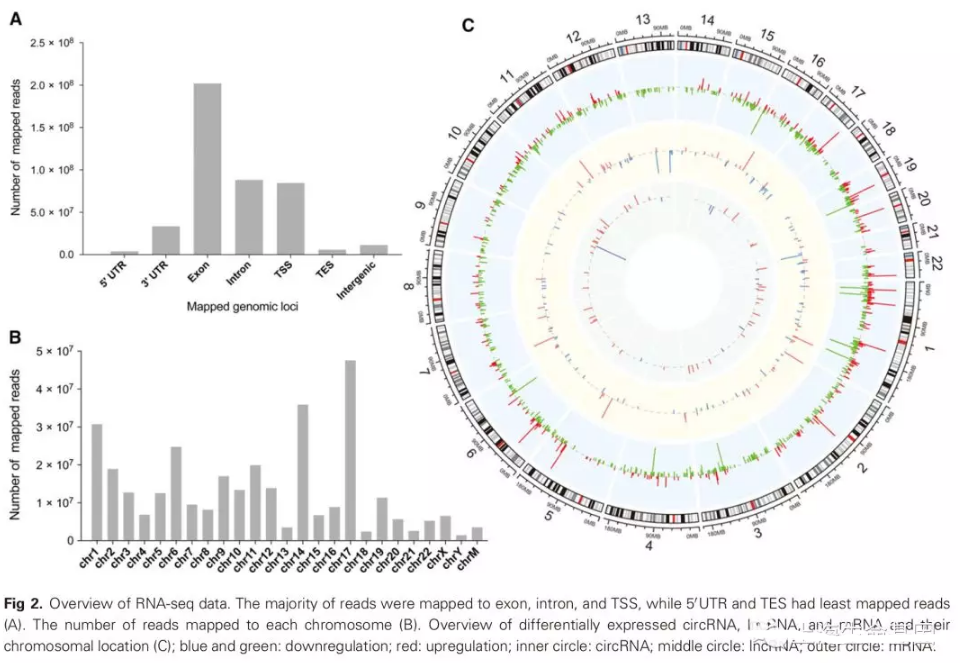
数据分析流程如Fig. 1。肺腺癌组织及癌旁组织提取total RNA,然后去除 rRNA。首先将reads比对到基因组分析mRNA, lncRNA的表达量,然后将 unmapped reads进行circRNA预测以鉴定circRNA表达谱,然后分析lncRNA, circRNA, mRNA之间的关系。最后,对差异表达的基因,进行功能富集分析,以了解其功能。大多数reads比对到exon, intron, and TSS,5’UTR的最少(fig. 2A)。Chr18,chr14,chr1比对到的reads数最多,同时有些reads比对到线粒体(fig. 2B)。figure 2C概括了lncRNA, circRNA, mRNA差异表达的情况。
2 编码基因表达谱
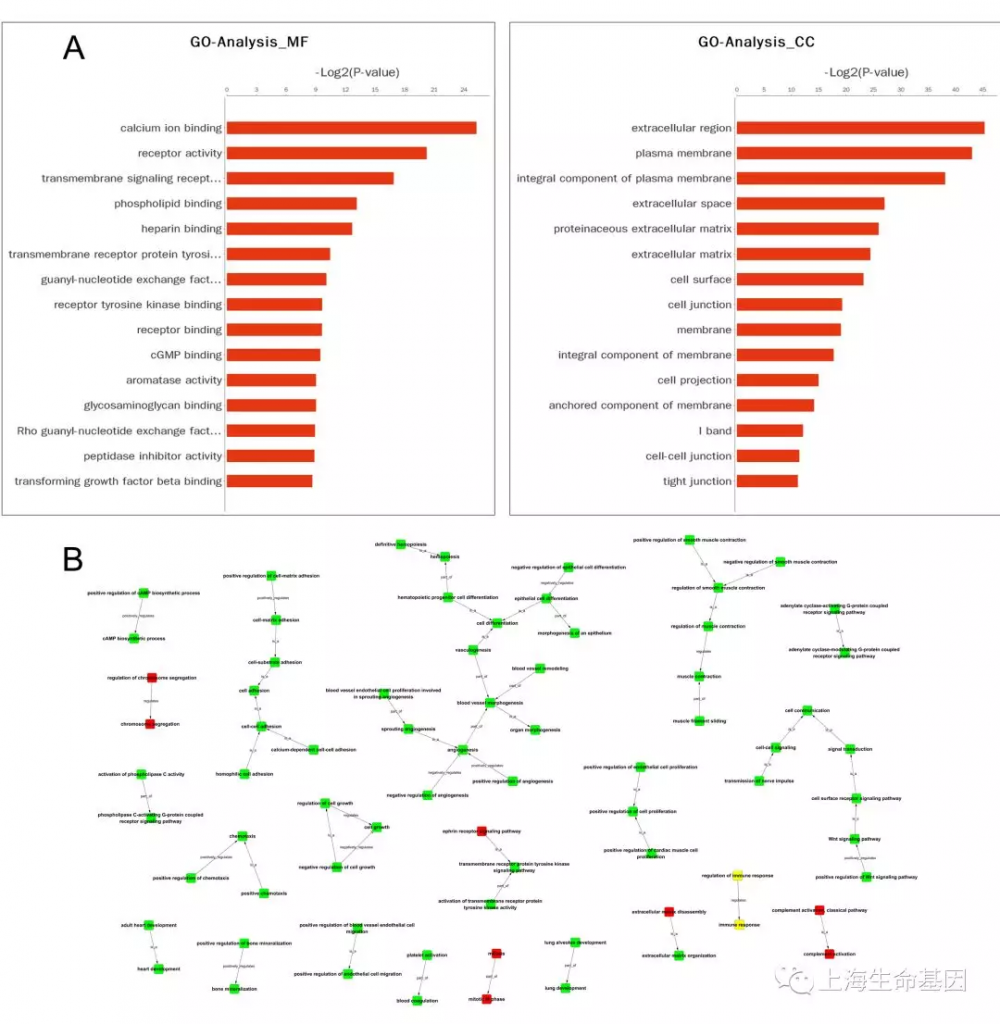
共检测到20384个mRNA, 1282个显著差异表达(Fig. 3A)。GO分析显示,差异表达基因主要涉及到分子功能如calciumion biding(钙离子结合), receptor activity(受体激活), transmembrane signaling receptor activity(跨膜信号受体激活), 及其他重要功能(Fig. S1A)。GO细胞组分分析主要包括,extracellular matrix (细胞外基质)and plasma membrane(质膜)(Fig. S1A)。GO生物学过程显著富集到cell adhesion(细胞黏着), angiogenesis(血管生成), extracellular matrix organization(胞外机制构成), and cell surface receptor signaling pathways(细胞表面受体通路)(Fig. 3B)。上述过程均与癌转移相关。对生物学过程进一步GO tree分析发现,细胞-细胞黏着,血管生成是核心过程(Fig. S1B)。KEGG显著富集到cGMP-PKG signaling pathway, cell adhesion molecules (CAMs), neuroactive ligand–receptor interaction(Fig. 3C)。Calcium signaling pathway, cell adhesion molecules, PI3K-Akt signaling pathway, and focal adhesion是核心通路(Fig. 3D)。
![]()
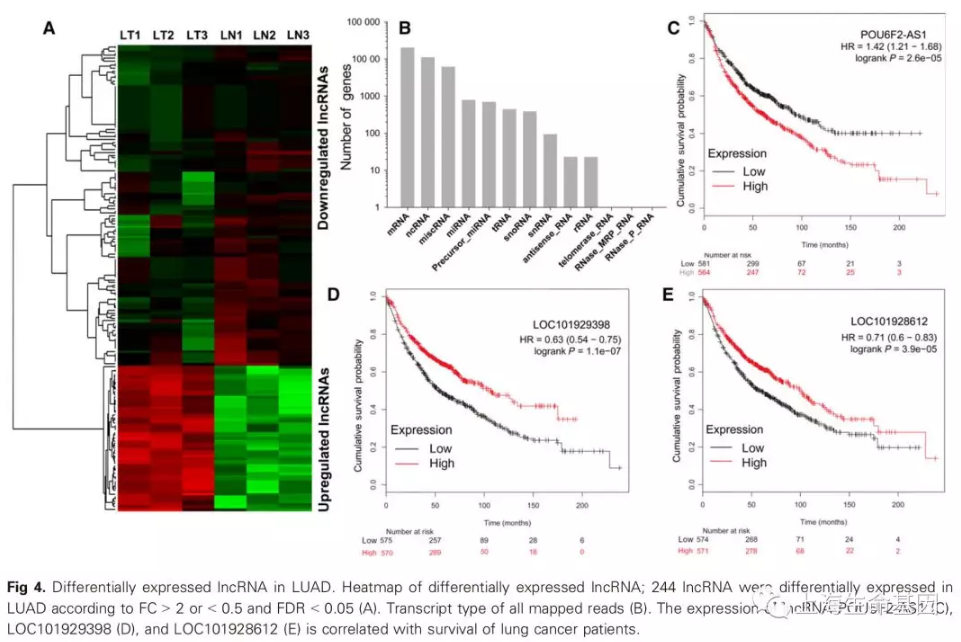
3 肺腺癌lncRNA表达
lncRNA共鉴定到19023个,显著差异的有244个(Fig. 4A; Table S3)。比对到转录组的reads类型如Fig. 4B, 可以看出ncRNA reads大概占据所有比对reads的三分之一。AFAP1-AS1, BLACAT1, and LOC101928245是上调最显著的lncRNA。FENDRR, LOC400406, and LINC00842是下调最显著lncRNA。我们组及其他研究人员曾报道AFAP1-AS1、BLACAT1在肺癌中扮演重要角色。FENDRR在肺癌中的作用也曾报道过。LncRNA的异常表达,常与临床特征,病人生存率密切相关。高表达的AFAP1-AS1、BLACAT1与肺癌较差预后密切相关。通过一些网上工具,我们发现POU6F2-AS1,
LOC101929398, and LOC101928612与肺癌的生存率相关(Fig. 4C-E)。提示lncRNA可能在肺癌中扮演重要角色。
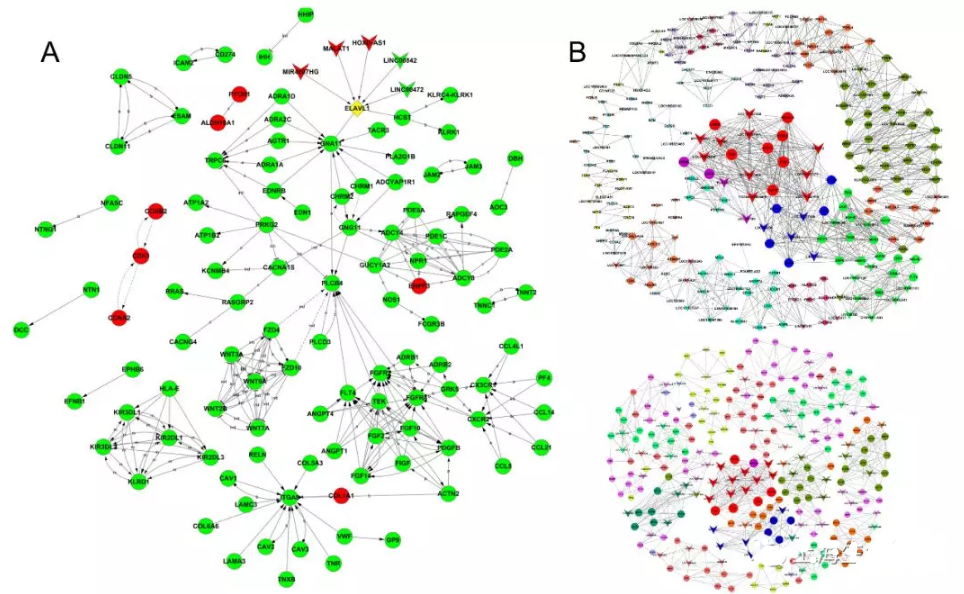
我们对lncRNA-protein关系进行预测,结果提示lncRNA与蛋白质ELAVL1(Fig. S2A)相关。构建lncRNA-mRNA网络进而预测lncRNA的生物学过程。可以发现肺癌的共表达网络与癌旁共表达网络是不同的。lncRNA BALCAT1, LOC101929398, and MYO16-AS1是关键的节点,而且拥有网络最高的度。考虑到BALCAT1 and LOC101929398与肺癌预后的关系,推测,网络中度较高的lncRNA,可能在肺癌发生过程起重要的功能。
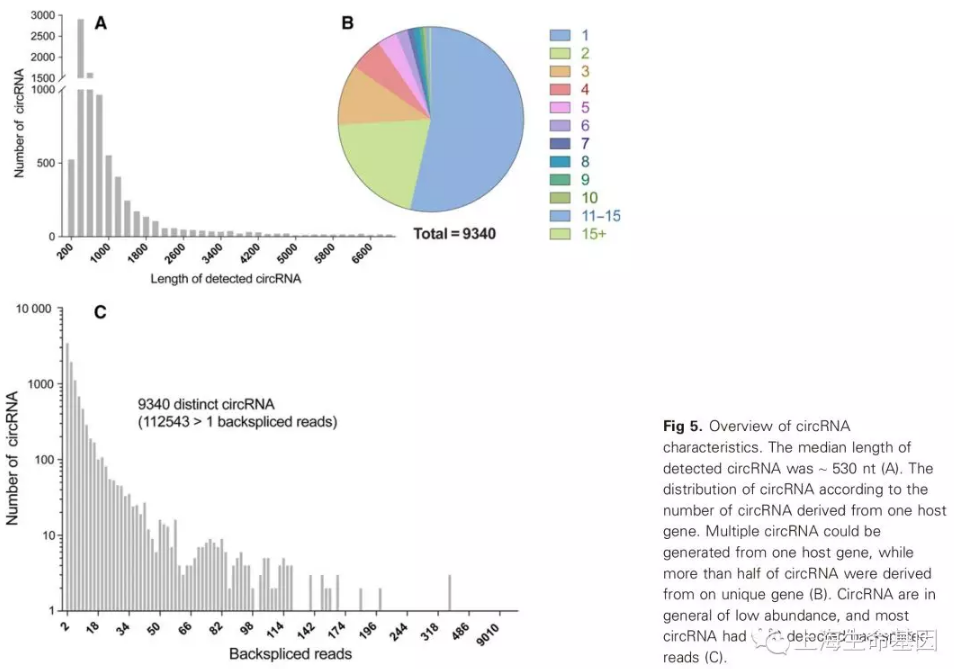
circRNA预测算法共鉴定到9340个circRNA(≥ 2 backspliced reads);与数据库报道的相比,5750个circRNA是已知的,3590个novel circRNA是本次研究鉴定到的。新鉴定到的circRNA,在Appendix S1。
大部分的circRNA长度<1000nt(6579个circRNA的长度小于1000nt),中值是530nt(Fig. 5A),与以往的报道一致。circRNA表达丰度要小于mRNA和lncRNA。鉴定到112543 backspliced reads,其中超过一半的circRNA的backspliced reads <10(Fig. 5B)。一个来源基因可以产生多个circRNA(Fig. 5C), MACF1可以产生多达26个circRNA。另外,许多基因只产生一个circRNA, 约有1/4的circRNA是其来源基因仅有的环状RNA产物(2307 of 9340 circRNA, Fig. 5C)。
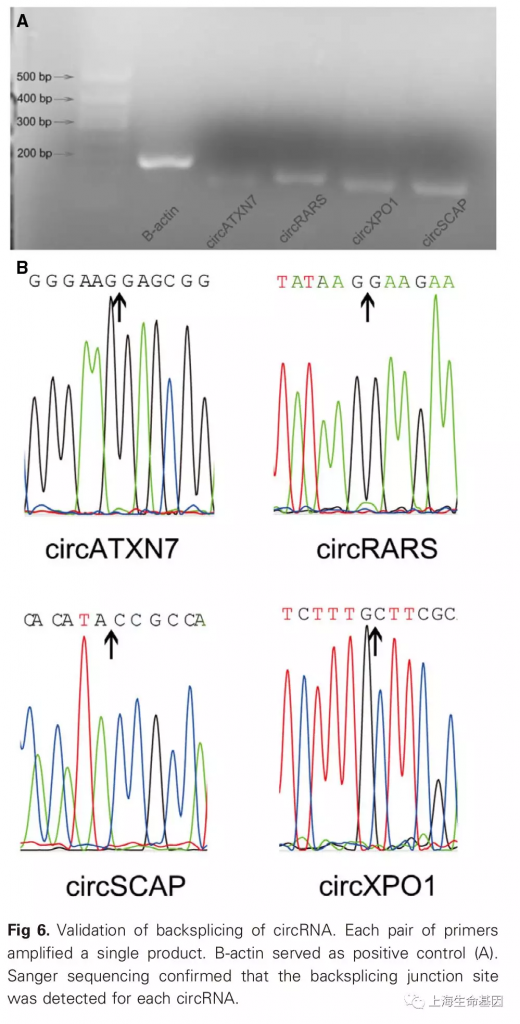
为了确定backspliced reads是否确实是环状的,而非普通转录本剪切的产物,对四个差异表达交大的circRNA,设计divergent primers(发散性引物),扩增A549 cell cDNA(Fig. 6A),使用Sanger测序确定其连接位点(Fig. 6B)。
Fold change (FC) >2 或 <0.5, flase discovery rate (FDR) <0.05, 共发现56个差异表达的circRNA(Table S4)。circRNA与mRNA共表达分析以预测circRNA潜在生物学功能。功能相似或位于相同通路的基因通常具有相似的表达模式。一些重要的癌基因与circRNA共表达,如Wnt3A, CDK1,and BUB1,说明这些circRNA可能参与肺癌的发生过程(Fig. S3)。
4 讨论
虽然发现了几十年,直到近些年,非编码RNA才受到广泛关注。肺癌的lncRNA表达谱也被广泛报道,但肺癌中circRNA的表达谱却研究的很少。本研究,我们使用RNAseq rRNA去除的方式深入分析了肺腺癌中的mRNA, lncRNA, circRNA表达。发现了244个差异表达的lncRNA,及3590个新的circRNA, 并有56个差异表达的circRNA。
对于circRNA形成,人们提出过几种机制。Bachmayr et al.发现,circRNA的表达丰度与增值呈负相关,推断circRNA更容易在非增值细胞中发生积累。在研究上皮间质转化时,Conn发现许多高丰度circRNA,受到剪切因子的调控,提示circRNA的形成,会受到RNA结合蛋白的调控。对于外显子来源circRNA, 互补序列,比如在两端内含子上Alu elements,上下游内含子会与蛋白质结合,促使circRNA环化。
本研究我们发现大部分的circRNA来源于前体mRNA外显子和内含子的环化,这与之前报道的大部分circRNA来源于编码序列的报道一致。对circRNA 及来源基因的数量分析,发现一个基因可以产生多个circRNA。我们观察到MACF1基因可以产生26个不同的circRNAs,之前有报道PTK2基因能够产生47个不同的circRNA。大部分circRNA的长度在200-800nt之间,与之前报道的circRNA中值的长度500nt一致。
众多证据表明,circRNA在多种生物学过程和疾病中发挥作用,在大部分功能中circRNA发挥microRNA海绵吸附作用。一个非常夸张的例子是,环状RNA CDR1as在脑组织中具有miR-7超过60个保守结合位点。为了预测高丰度表达circRNA的功能,我们构建circRNA-mRNA网络。许多癌基因与circRNA共表达,比如Wnt3a, and CDK1。由此推断circRNA或许参与这些生物学过程。
对真核RNA的研究,通常会先进行polyA富集。这会失去一部分没有polyA尾的lncRNA。本研究我们使用rRNA去除RNAseq建库方式,来研究肺腺癌lncRNA表达谱,这有助于我们检测到没有polyA尾的lncRNA。据我们所知,这是第一次使用rRNA去除的RNAseq技术研究肺腺癌。AFAP1-AS1, BLACAT1, and FENDRR是肺腺癌差异最明显的lncRNA, 这与我们之前的工作和别人的报道一致,说明RNAseq技术是比较可靠的。构建lncRNA-mRNA共表达网络来预测lncRNA的功能。在网络中,BALCAT1 and LOC101929398拥有的度最高,位于网络的中心。BALCAT1之前报道是肺癌的一个致癌基因。Kaplan–Meier曲线显示高表达的LOC101929398肺癌差预后相关。这些说明,LOC101929398可能是一个原癌基因。通过共表达网络,我们可能鉴定到在肺腺癌中发挥作用的lncRNA。
5 结论
通过rRNA去除的RNAseq技术,我们分析了肺腺癌mRNA, lncRNA, circRNA表达谱。AFAP1-AS1,BLUADAT1,LOC101929398是上调最明显的lncRNA。我们检测到9340个circRNA,包括3570个新的circRNA,说明circRNA在肺腺癌中广泛表达。
6 研究思路整理
研究材料:9对肺腺癌及癌旁样品,提取RNA后,混成3个癌组织样品,3个癌旁组织样品。
研究过程:
1 、使用rRNA去除的方式建库,获得mRNA, lncRNA, circRNA的信息。
2、 对原始下机数据reads去除接头序列,去除“N”超过5%的序列,去除碱基质量值<13含量大于20%的reads。MAPSPLICE 软件 (v2.1.8) 将clean reads比对到人基因组(version: GRCh37),为了获得更多的可变剪切转录本,设置参数为(–s 22 –p 12 –ins 6 –del 6 –noncanonical)。使用acfs流程https://code.google.com/p/acfs/鉴定circRNA。
3 、差异表达分析使用DESeq算法过滤差异表达的基因,FC和FDR作为筛选标准,标准如下(a) FC > 2 or < 0.5; (b) FDR< 0.05。
4 、GO分析使用Fisher’s exact test计算p-value,使用FDR值对p-value进行校对。
5 、使用KEGG数据库进行通路分析以鉴定有意义的通路,同样使用Fisher’s exact test计算p-value,使用FDR值对p-value进行校对。FDR<0.05的认为有意义。
6 、GO tree是一种有向无环图,条目之间彼此关联,可以提供友好的数据整理和展示。我们挑选了有意义的GO条目(P-value<0.01)来构建GO tree来了解实验影响的功能。
7、 KEGG数据库包含代谢、膜转运、信号转导、细胞周期通路。我们选择富集到的生物学过程的基因,使用CYTOSCAPE(version 3.2.1)进行通路图形化展示。
8、 共表达网络,对mRNA-lncRNA, mRNA-circRNA根据表达量相关性分别构建共表达网络。
9 、使用LncRNA-protein关系构建 lncRNA-protein关系 网络。
结果展示:
1 、本次测序数据分析流程(Fig. 1),reads比对到基因组不同区域的统计饼图(fig. 2A),reads比对到染色体数量统计柱状图(fig. 2B),lncRNA, mRNA, circRNA差异分布circos图(figure 2C)。
2 、基因表达分析:差异表达基因的热图(Fig. 3A),差异表达基因GO富集生物学过程柱状图((Fig. 3B),分子功能柱状图(Fig. S1A),细胞组分柱状图(Fig. S1A),GO tree图(Fig. S1B),KEGG通路柱状图(Fig. 3C),通路网络图(Fig. 3D)。
3 、lncRNA表达分析:差异表达lncRNA热图(Fig. 4A),lncRNA表达列表(Table S3),比对到基因组区域的统计柱状图(Fig. 4B),POU6F2-AS1, LOC101929398, LOC101928612生存曲线(Fig. 4C–E),lncRNA-protein网络图(Fig. S2A),lncRNA-gene共表达网络图(Fig. S2B)。
4 、circRNA表达分析:统计circRNA数量,差异数量,新circRNA数量(表Appendix S1),长度分布柱状图(Fig. 5A),来源基因数量饼图(Fig. 5B),连接点reads数量柱状图(Fig. 5C)等信息,设计引物扩增circRNA电泳图(Fig. 6A),Sanger测序验证circRNA连接点峰图(Fig. 6B),circRNA差异表达列表(Table S4),circRNA mRNA共表达网络(Fig. S3)。
另:材料方法中附样品临床资料(Table S1),circRNA引物(Table S2)。

来第一个抢占沙发评论吧!